
| Rev Esp Endocrinol Pediatr 2019;10(2):41-47 | Doi. 10.3266/RevEspEndocrinolPediatr.pre2019.Jul.495 | |||||||||
| Tratamiento con GH en pacientes con alteraciones del gen SHOX: nuestra experiencia | |||||||||
| GH treatment in patients with alterations of SHOX gene: our experience | |||||||||
| Sent for review: 14 Jan. 2019 | Accepted: 24 Jul. 2019 | Published: 30 Dec. 2019 | |||||||||
| María Aiguabella Font1, Arancha Escribano Muñoz2, José María Martos Tello2, Mª José Romero Egea2 | |||||||||
| 1 Servicio de Pediatría. Hospital General Universitario Santa Lucía. Cartagena, Murcia 2Sección de Endocrinología Pediátrica, servicio de Pediatría. Hospital Clínico Universitario Virgen de la Arrixaca. Murcia | |||||||||
| Correspondence:María Aiguabella Font, Servicio de Pediatría, Hospital General Universitario Santa Lucía, Cartagena, Murcia E-mail: maria.aiguabella@gmail.com | |||||||||
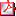 Tabla 1 - Mutaciones genéticas identificadas, necesidad o no de tratamiento con GH, sexo y necesidad o no de tratamiento con análogos de GnRH de nuestros pacientes Tabla 1 - Mutaciones genéticas identificadas, necesidad o no de tratamiento con GH, sexo y necesidad o no de tratamiento con análogos de GnRH de nuestros pacientes | |||||||||
| Tabla 2 - Resultados | |||||||||
| |||||||||
INTRODUCCIÓN La talla baja constituye uno de los principales motivos de consulta tanto en las consultas de Atención Primaria como en las consultas especializadas de Endocrinología Pediátrica, representando hasta el 30% de las consultas en algunas series (1). Aunque es complejo establecer la normalidad de una talla, puesto que el crecimiento es un proceso dinámico e influenciado por múltiples factores -genéticos, hormonales, ambientales-, se ha establecido la definición de talla baja como: una talla menor a -2 desviaciones estándar (DE) para niños del mismo sexo, edad y etnia; una talla entre ±2 DE para la población general pero menor a -2 DE por debajo del carril de crecimiento correspondiente a su talla diana; una expectativa de talla adulta más de 2 DE por debajo a su talla diana o una velocidad de crecimiento inferior a -1 DE para edad y sexo mantenida más de 2-3 años (2). Esta definición, sin embargo, difiere ligeramente de los criterios auxológicos que deben cumplir los pacientes candidatos al tratamiento con GH fijados por el ministerio de sanidad español en el año 2008, entre los que se incluyen: una talla menor a -2 DE o por debajo de 1 DE de la talla media parental y, en su caso, predicción de talla adulta inferior a la talla genética en más de 1 DE; una velocidad de crecimiento por debajo del percentil 10 para su correspondiente edad ósea, mantenida durante un mínimo de 6 meses y un retraso de la maduración ósea en más de 1 año con respecto a la edad cronológica (3). Dentro de la definición de talla baja, se acepta que una talla <-3 DE o una talla desproporcionadamente baja para la talla familiar es más sugestivo de que se trate de una talla baja patológica frente a una talla baja idiopática o una variante de la normalidad, encontrándose hasta en un 58% de estos casos una causa orgánica que la justifique (4). Las múltiples causas de talla baja patológica las clasificamos en dos grandes grupos: talla baja proporcionada, dentro de las cuales distinguimos aquellas de origen prenatal de las de origen postnatal, y talla baja desproporcionada, dentro de las cuales se incluyen las displasias esqueléticas. La deficiencia de SHOX puede condicionar diferentes fenotipos clínicos, desde formas más graves como la displasia mesomélica de Langer hasta formas con menor afectación como la discondrosteosis de Leri-Weill o formas inicialmente diagnosticadas como talla baja idiopática (5). El gen SHOX y su relación con los trastornos del crecimiento fue descrito por primera vez en el año 1997, de forma paralela por dos grupos de investigadores (6 y7) en la búsqueda de factores condicionantes de la talla baja observada en el síndrome de Turner. En la actualidad, se estima que puede afectar a 1 de cada 2000-4000 niños, suponiendo la causa monogénica más frecuente de talla baja. Desde su descripción, y sobre todo en la última década, su diagnóstico ha ido adquiriendo cada vez más relevancia clínica. El gen SHOX se localiza en la región pseudoautosómica 1 (PAR1) localizado distalmente en los brazos cortos de los cromosomas X e Y. Los estudios inmunohistoquímicos demuestran que la expresión de SHOX está vigente desde la semana 12 de la gestación, tanto en la zona de reserva, como las proliferativa e hipertrófica del cartílago de crecimiento (8). Actuaría a nivel de la zona de reserva impidiendo la diferenciación del condrocito y su progresión hacia las zonas proliferativa e hipertrófica. Las localizaciones principales serían el primer y segundo arco branquiales y extremo distal de los miembros (muñeca, radio, cúbito, fémur distal y tibia), que condicionarían ulteriormente parte de la clínica observada en estos pacientes (9). Fue a raíz de los resultados obtenidos por Blum et al (10) cuando se aceptó como nueva indicación para el tratamiento con GH. Posteriormente se han ido publicando series más amplias que reflejan globalmente un beneficio en talla derivado del tratamiento con hormona de crecimiento y sin efectos secundarios destacables. Así pues, tanto por su frecuencia como por su beneficio terapéutico, su identificación resulta esencial (11). PACIENTES Y MÉTODOS Es un estudio descriptivo y retrospectivo en el cual revisamos las historias clínicas de todos nuestros pacientes en seguimiento en consultas externas de Endocrinología Pediátrica de nuestro hospital terciario desde Mayo de 2008 hasta marzo de 2017 en los que se identificó una alteración del gen SHOX y sus secuencias reguladoras mediante MLPA y que han recibido tratamiento con GH. Desde 2015 y en el marco del proyecto Crecemos (12), además de MLPA, se realiza la búsqueda de mutaciones puntuales y pequeñas inserciones/deleciones (indels) en la región codificante del gen SHOX mediante HRM y secuenciación Sanger. Excluimos del análisis aquellos pacientes que presentaban una talla normal o que no cumplían los criterios auxológicos para la financiación del tratamiento, y que, por tanto, no han recibido tratamiento con GH. La dosis de GH recibida oscila entre 0,036 y 0,050 mg/kg/día. Todos los datos auxológicos fueron recogidos utilizando el mismo tallímetro y por personal entrenado en técnicas antropométricas. Como estándares de crecimiento se utilizaron los datos del estudio español de crecimiento de 2010 (13). Recogimos los datos de aquellas variables que podrían influir en la respuesta al tratamiento: edad cronológica al inicio del tratamiento, sexo, talla diana, talla al inicio, edad ósea al inicio, expectativa de talla adulta al inicio del tratamiento, velocidad de crecimiento (VC) al inicio, estadio de Tanner al inicio del tratamiento, proporciones corporales (envergadura y talla sentado), fenotipo clínico y tipo de mutación identificada. Y las comparamos con aquellas variables indicadoras de respuesta al tratamiento: ganancia de talla al año, a los dos años y al término del tratamiento; ganancia de velocidad de crecimiento al año y a los dos años y ganancia de expectativa de talla al año, a los dos años y al término del tratamiento. Para el análisis estadístico empleamos el programa STATA y las variables se relacionaron entre sí mediante análisis de regresión lineal, teniendo también en cuenta aquellas variables que pudieran actuar como factor de confusión. Dada la naturaleza y diseño del estudio (observacional, retrospectivo y con nulo riesgo para los pacientes) el Comité Ético de nuestro hospital concedió a los autores la exención de obtener consentimiento informado, comprometiéndose a garantizar la confidencialidad de los datos obtenidos. RESULTADOS En nuestra serie hemos identificado 8 mutaciones distintas del gen SHOX en 18 pacientes (tabla 1). La más frecuente (identificada en 6 pacientes) es la deleción recurrente de 47,5 kb de la región pseudoautosómica PAR1, que incluye 3 zonas reguladoras de la expresión del gen SHOX (CEN7, CEN8 y CEN8/9). El resto son deleciones amplias o parciales en heterocigosis y sólo en un caso hemos encontrado una mutación puntual y en otro una duplicación en heterocigosis. No hemos encontrado relación estadísticamente significativa entre el tipo de mutación y la afectación de la talla o la necesidad o no de recibir tratamiento con GH. Seis de los pacientes no cumplen los criterios auxológicos para iniciar tratamiento (50% mujeres, 50% varones). Ocho de los pacientes (63% mujeres, 37% varones) están en tratamiento con GH actualmente. Cuatro de los anteriores también han recibido tratamiento con análogos de GnRH. Uno de nuestros pacientes, varón, se encontraba a la espera de ser valorado por el Comité de GH de la comunidad autónoma para decidir iniciar tratamiento en el momento de recogida de los datos. Sólo tres pacientes han completado el crecimiento (tabla 1). De los cuatro pacientes que han recibido tratamiento con análogos de GnRH sólo uno de ellos ha completado el crecimiento. No hemos encontrado diferencias estadísticamente significativas en cuanto a ganancia de talla entre los pacientes que habían recibido tratamiento exclusivo con GH y a los que se asoció análogos de GnRH. Los 11 pacientes que han recibido tratamiento (73% mujeres, 27% varones) presentaban una edad media al inicio del tratamiento de 9 años 8 meses (± 1 año 6 meses) (tabla 2). Todos presentaban una edad ósea adelantada al inicio del tratamiento, con un ratio edad ósea (EO)/edad cronológica (EC) ≥1 y un pronóstico de talla adulta (PTA) patológico previo al tratamiento (<-2DE). De los 11 pacientes, 7 son los que han completado un año de tratamiento. Este grupo presentaba una talla media inicial en -1,95(±0,55) DE; con una expectativa de talla adulta en -3,10(±0,94) DE. En ellos, al finalizar el primer año de tratamiento hemos observado: una ganancia de +0,84(±0,41) DE de talla y una ganancia de +1,54(±0,68) DE de expectativa de talla adulta. La velocidad de crecimiento (VC) media de +3,17(±1,66) DE, suponiendo una ganancia de +4,03(±2,44) DE respecto al inicio del tratamiento, comparándola con la VC en el año previo (tabla 2). De los 11 pacientes, son 5 los que han completado dos años de tratamiento. Este grupo presentaba una edad cronológica media de 9 años 6 meses (±1 año 6 meses) al inicio del tratamiento; una talla media inicial de -1,91(±0,64) DE y una expectativa de talla adulta de -2,88(±0,80) DE. En ellos hemos observado: una talla media a los 2 años de -0,98(±0,81) DE, suponiendo una ganancia +0,92(±0,60) DE respecto al inicio; una expectativa de talla al segundo año de -1,27(±0,97) DE, representando una ganancia +1,61DE (±1,06) DE respecto al inicio. La VC media fue de +0,77(±1,44) DE el 2º año (tabla 2). Los tres pacientes que han finalizado el crecimiento (paciente 4, paciente 16 y paciente 18 en tabla 1) tenían una edad cronológica al inicio del tratamiento de 8 años y 3 meses de media (± 10 meses), presentan una talla final de -1,65(±0,26) DE, con una ganancia media de talla con respecto al inicio del tratamiento de +0,47(±0,21) DE (tabla 2). Teniendo en cuenta que la expectativa de talla al inicio del tratamiento era de -2,62(±0,40) DE supone una mejoría de casi en 1DE con respecto a la talla final. En el análisis estadístico que relaciona las variables respuesta (VC y ganancia de talla el 1º y el 2º año) entre sí y con aquellas que podrían influenciar en la respuesta al tratamiento (sexo, tipo de mutación, talla al inicio, edad al inicio, PTA al inicio, VC al inicio y afectación de segmentos corporales), encontramos una relación estadísticamente significativa entre: mayor ganancia de talla el 2º año con respecto a mayor ganancia de talla el 1º año (p = 0,006); mayor ganancia de talla el 1º año respecto a menor edad (p = 0,009) y mayor ganancia de talla el 1º año respecto a menor talla al inicio (p = 0,037). Se encuentra en el límite de la significación estadística la relación entre mayor ganancia de VC el 1º año y peor PTA al inicio (p = 0,05). No encontramos relación estadísticamente significativa con el resto de variables estudiadas. DISCUSIÓN Y CONCLUSIONES En nuestra serie observamos una respuesta global al tratamiento: una media de Δ-SDS-talla de +0,84 al año de tratamiento, de +0,92 a los dos años y de +0,47 al completar el crecimiento y una media de Δ-SDS-velocidad de crecimiento de +4,03 al año de tratamiento, comparable con los resultados descritos en la bibliografía (11), aunque sería necesario incluir a más pacientes y con mayor tiempo de seguimiento para aumentar la potencia del estudio. Por otro lado, muchos de los estudios previamente publicados (11) consisten en series de casos de pacientes con discondrosteosis de Leri-Weill y en algunos de ellos la ganancia de talla descrita con dosis similares de GH es mayor. En nuestro estudio, se trata de una serie de casos de pacientes con diferentes fenotipos clínicos: desde pacientes catalogados inicialmente como talla baja idiopática con algunos rasgos propios de la deficiencia del gen SHOX, tanto clínicos como radiológicos, hasta pacientes con fenotipo Leri-Weill. En cualquier caso, el escaso tamaño muestral dificulta la identificación de relación estadísticamente significativa entre las variables estudiadas, ya que es probable que a mayor afectación clínica la ganancia de talla con el tratamiento sea mayor. Por otra parte, tres de nuestros pacientes que han requerido tratamiento con GH presentan una talla diana patológica, probablemente por la afectación de alguno de los padres. Esto, posiblemente, influya de forma negativa en cuanto a la ganancia de talla final. Los datos recogidos muestran en nuestra serie de pacientes una predominancia femenina en cuanto a la necesidad de tratamiento con GH, aunque no hemos observado diferencias estadísticamente significativas entre la afectación de la talla al inicio del tratamiento y el sexo, lo cual también concuerda con los resultados descritos en la bibliografía hasta la fecha (11). Esto podría explicarse dado el papel regulador de SHOX más aceptado en la actualidad, que es el de promotor del crecimiento lineal de las extremidades, funcionando como represor de la fusión y maduración de la fisis, contrarrestando el efecto de los estrógenos. Este hecho también explicaría el predominio puberal observado en la clínica de los pacientes con defectos en gen SHOX. Hemos observado que la edad al inicio de tratamiento con GH sí se relaciona de forma indirectamente proporcional con una mejor respuesta al tratamiento. La identificación precoz de estos pacientes se ve dificultada por el predominio puberal de la clínica y por el hecho de que la desproporcionalidad corporal se va acentuando a medida que se completa el crecimiento. De igual modo que en estudios previos (14), no hemos observado diferencias fenotípicas en cuanto al mecanismo productor de la alteración genética. CONFLICTOS DE INTERESES Los autores no declaran conflicto de intereses alguno en relación con este artículo. | |||||||||
| References | |||||||||
1. Sevilla Ramos, M.P.; Alija Merillas, M.J.; López Andrés, N. Diagnóstico de talla baja en consulta de endocrinología pediátrica de un hospital provincial secundario. Rev Esp Endocrinol Pediatr 2014 ;5(2):9-17. doi: https://doi.org/10.3266/RevEspEndocrinolPediatr.pre2014.May.213. 2. Pozo Román, J. Crecimiento normal y talla baja. Pediatr Integral 2015; XIX (6): 411.e1-411.e23. 3. Comité asesor para la hormona de crecimiento del ministerio de sanidad y consumo. Criterios para la utilización racional de la hormona de crecimiento en niños. Mayo 2008. Disponible en: https://www.mscbs.gob.es/profesionales/farmacia/pdf/criteriosHCninos020908.pdf. 4. Voss LD, Mulligan J, Betts PR, Wilkin TJ. Poor growth in school entrants as an index of organic disease: the Wessex growth study. BMJ. 1992;305(6866):1400-2.[Pubmed] 5. Rosell Andreo, J. Talla baja por alteraciones del gen SHOX. Rev Esp Endocrinol Pediatr 2011;2 Suppl(1):25-26. doi: https://doi.org/10.3266/RevEspEndocrinologPediatr.pre2011.Apr.46. 6. Rao E, Weiss B, Fukami M, Rump A, Niesler B, Mertz A, et al. Pseudoautosomal deletions encompassing a novel homeobox gene cause growth failure in idiopathic short stature and Turner syndrome. Nat Genet. 1997;16(1):54-63.[Pubmed] 7. Ellison JW, Wardak Z, Young MF, Gehron Robey P, Laig-Webster M, Chiong W. PHOG, a candidate gene for involvement in the short stature of Turner syndrome. Hum Mol Genet. 1997;6(8):1341-7.[Pubmed] 8. Munns CJF, Haase HR, Crowther LM, Hayes MT, Blaschke R, Rappold G, et al. Expression of SHOX in human fetal and childhood growth plate. J Clin Endocrinol Metab. 2004;89(8):4130-5.[Pubmed] 9. Clement-Jones M, Schiller S, Rao E, Blaschke RJ, Zuniga A, Zeller R, et al. The short stature homeobox gene SHOX is involved in skeletal abnormalities in Turner syndrome. Hum Mol Genet. 2000;9(5):695-702.[Pubmed] 10. Blum WF, Crowe BJ, Quigley CA, Jung H, Cao D, Ross JL, et al. Growth hormone is effective in treatment of short stature associated with short stature homeobox-containing gene deficiency: Two-year results of a randomized, controlled, multicenter trial. J Clin Endocrinol Metab. 2007;92(1):219-28.[Pubmed] 11. Barreda-Bonis, C.; González Casado, I. Tratamiento con GH en alteraciones del gen SHOX. Rev Esp Endocrinol Pediatr 2016;7 Suppl(1):9-23. doi: https://doi.org/10.3266/RevEspEndocrinolPediatr.pre2016.Apr.338. 12. M. Aza Carmona, A. Belinchon Martinez, C. de la Torre Gómez, A.C. Barreda Bonis, I. González Casado, K.E. Heath. “Proyecto Crecemos”: resultados de 48 meses del análisis del gen SHOX. Rev Esp Endocrinol Pediatr 2018; 9 (Suppl 1); 67. doi: https://doi.org/10.3266/RevEspEndocrinolPediatr.pre2018.Apr.464. 13. Carrascosa, A.; Fernández, J.M.; Fernández, A.; López-Siguero, J.P.; López, D.; Sánchez, E. et al. Estudios españoles de crecimiento 2010. Disponible en: http://www.seep.es/privado/documentos/Publicaciones/Estudios_Españoles_de_Crecimiento_2010.pdf. 14. Munns CFJ, Glass IA, Flanagan S, Hayes M, Williams B, Berry M, et al. Familial growth and skeletal features associated with SHOX haploinsufficiency. J Pediatr Endocrinol Metab. 2003;16(7):987-96.[Pubmed] | |||||||||
 |
 |
 |
 |
 |
 Print
Print View / Download PDF
View / Download PDF  English version
English version Comments
Comments Add Favorites
Add Favorites