
| Rev Esp Endocrinol Pediatr 2011;2 Suppl(1):165-174 | Doi. 10.3266/RevEspEndocrinologPediatr.pre2011.May.61 |
| Miscelánea |
| Sent for review: 2 May. 2011 | Accepted: 3 May. 2011 | Published: 3 May. 2011 |
| Congreso SEEP |
| Correspondence: Congreso SEEP E-mail: seep@seep |
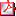 Tabla - P1d2-3-132_012_prolactinomas Tabla - P1d2-3-132_012_prolactinomas |
| Tabla - P1d2-3-137_012_Síndrome de Morsier de presentación neonatal |
 Figura - P1-d2-3-063 Figura - P1-d2-3-063 |
| Figura - P1-d2-3-116 |
|
|
P1/d2-3-129 SÍNDROME OSTEOPOROSIS-PSEUDOGLIOMA: EVOLUCIÓN CLÍNICA TRAS TRATAMIENTO CON BIFOSFONATOS Y HORMONA DE CRECIMIENTO. M.G. Bueno Lozano(1), J. Fleta Zaragozano (1), Bueno Lozano O (1), Ramos Fuentes F (2), I. Bueno Martínez (2), JM. Garagorri Otero (3).
(1) Servicio de Pediatría, (2) Unidad de Genética, (3) Unidad de Endocrinología Pediátrica. Hospital Clínico Universitario Lozano Blesa, Zaragoza.
Introducción: El Síndrome Osteoporosis-Pseudoglioma (MIM 259770), es una enfermedad rara y poco conocida con transmisión autosómica recesiva. Clínicamente cursa con ceguera de aparición precoz y fragilidad ósea que da lugar a fracturas múltiples desde la primera infancia. La alteración genética se relaciona con mutaciones con pérdida de función del gen LRP5 (11q13) que codifica dicho receptor, íntimamente implicado en los procesos de formación ósea.
Caso Clínico: Niña de 10,4 años de edad, procedente de Rumanía que consulta por cuadro clínico consistente en ceguera bilateral detectada al tercer mes de vida y fracturas múltiples de huesos largos desde su primer año. Tercera hija de una pareja sana, no consanguínea, que refiere la existencia de dos primos hermanos invidentes en familia materna así como otro caso similar en familia paterna. Exploración física: Peso: 23,5 Kg (< P3, a -2,03 s.d.s. ); Talla: 110 cm (
Pruebas complementarias: Calcemia: 10,3 mg/dL; Fosforemia: 5 mg/dL; Fosfatasa alcalina: 221 U/L; PTH: 43,4 pg/mL , 25(OH) D3: 28 ng/mL osteocalcina: 17 ng/mL. Función renal, aminoácidos y mucopolisacáridos en sangre y orina: normales. Mapa óseo: Osteopenia generalizada, huesos largos de menor calibre, platiespondilia con aplastamiento de cuerpos vertebrales y deformidad de caja torácica. Epífisis no afectadas. No alteraciones en pelvis ni cráneo. DMO(L1-L4): 0,378 g/cm2, a -4,3 s.d.s. para edad. Ecografía de órbitas: posibles restos de vítreo organizados. Cariotipo 46,XX; ADN(PCR): deleción c.2409-2503+79del174 en el gen LRP5.
Tratamiento: Ciclos de Pamidronato intravenoso (1mg/Kg/día, 3 días, cuatrimestral), Calcio (800 mg/día), Vitamina D3 (400U/día) y GH (0,2 mg/kg/semana). Tras el primer año de tratamiento, se ha observado una mejoría en la densidad mineral ósea, sin efectos secundarios reseñables.
Comentarios: El correcto funcionamiento del receptor LRP5 es esencial en la formación ósea. La mutación anteriormente descrita se ha relacionado con esta enfermedad (Ai M, 2005). Mutaciones menos graves del mismo gen se han relacionado con las formas graves de enfermedad osteoporótica en el adulto.
P1/d2-3-130 SINDROME DE MARFAN NEONATAL. L. Olivares Sánchez, R. Briones Pascual, P. Cid Galache, R. Pérez Iáñez, S. Broncano Lupiáñez y JM.
Servicio de Pediatría. Hospital de Motril, Motril.
El síndrome de Marfan (SM) es una enfermedad del tejido conectivo que afecta fundamentalmente a los sistemas cardiovascular, esquelético y ocular. Herencia autosómica dominante. Hasta un 25% de los casos pueden ser mutaciones de novo. Se asocia mayoritariamente a mutaciones en el gen FBN1 (15q21), que codifica la fibrilina 1, glicoproteína de las microfibrillas extracelulares. Tiene una gran variabilidad de expresión, por lo que su diagnóstico puede resultar difícil en casos no familiares esporádicos. Su diagnóstico en los primeros 3 meses de vida se denomina SM Neonatal, que asocia defectos cardíacos importantes (80% casos), contracturas congénitas (64%) y alta mortalidad neonatal (14%).
Se presenta el caso de un recién nacido varón, pretérmino (35 semanas), con peso 2.570 g, Talla 48, PC 35.5 cm y rasgos dismórficos (fenotipo Marfan): dolicocefalia, frente olímpica, ojos hundidos, retrognatia, tórax campaniforme con pectum excavatum, extremidades largas y delgadas con limitación global a la extensión y aracnodactilia marcada. Tono muscular y reflejos normales. Soplo sistólico 3/6. Distrés respiratorio (Silverman 4).
Embarazo sin incidencias. Padres sanos. No consanguíneos. No diagnóstico prenatal.
Exámenes complementarios: Ecocardiografía (3er día de vida): prolapso valvular mitral con insuficiencia mitral leve y dilatación de aorta ascendente a nivel de los senos de Valsalva. Examen oftalmológico: miopía, sin evidencia de subluxación del cristalino. Ecografía cerebral y RMN cerebral: normales.
Estudio genético (gen FBN1): mutación c.3143T>C en heterocigosis, (p.ILE1048Thr), cambio asociado al SM, en este caso de presentación neonatal. Está pendiente el estudio genético familiar. Actualmente tiene 2 años. Peso 11.3 kg (-0.92DS); Talla 93 cm (+2.1DS). Sigue tratamiento con atenolol y losartan, con progresión de la dilatación de raíz aórtica y precisa ortesis de columna y piernas.
Comentarios: El SM neonatal plantea el diagnóstico diferencial con el síndrome de Beals, producido por mutaciones del gen FBN2, cuyos hallazgos clínicos son muy similares pero cuya historia natural tiende a una mejoría gradual de las limitaciones articulares.
P1/d2-3-131 AFECTACIÓN ENDOCRINOLÓGICA TRAS TRAUMATISMOS CRANEOENCEFÁLICOS (TCE) EN LA INFANCIA. A. Salomon Estébanez(1), E. Morteruel ArizKuren (1), G. Grau Bolado (2), A. Rodríguez Estevez (2), A. Vela de Sojo (2), I. Rica Etxebarria (2).
(1) UCIP. Hospital de Cruces; (2) Endocrinología Infantil. Hospital de Cruces, Barakaldo. Vizcaya.
Los TCE son una causa de fallo hipotálamo-hipofisario. Estudios en adultos refieren una incidencia de disfunción hormonal del 30-70% tras TCE moderados-graves tanto en el periodo inicial como en fases posteriores. Los ejes más frecuentemente implicados son el somatotropo y el gonadal. En niños apenas existen estudios a pesar de la posible repercusión en el crecimiento y desarrollo puberal.
Objetivos: Estudiar la prevalencia de disfunción hipofisaria en niños con TCE. Conocer si existe relación entre gravedad del TCE y posible afectación endocrinológica.
Pacientes y Métodos: Estudio prospectivo de función endocrina en niños ingresados en UCIP por TCE con fractura de cráneo y/o sangrado intracraneal, entre 2004 y 2009. La severidad del traumatismo (grave, moderado o leve) se valora con la escala de Glasgow, la clínica y la imagen. Las secuelas se agrupan según la escala de secuelas de Glasgow (GOS). Se realiza una consulta endocrinológica valorando clínica, crecimiento y pubertad. Se determinan en analítica basal: T4 libre, TSH, PRL, cortisol, ACTH, estradiol o testosterona, LH, FSH, IGF-I e IGFBP3, iones en plasma y osmolaridad en plasma y en orina.
Resultados: - Se incluyen 34 pacientes: edad media de 3.8±3.7 años en el momento del TCE y 61.8% varones. La causa más frecuente de TCE fueron las caídas (63.3%). La gravedad del TCE fue: 64.7% leve, 14.7% moderado y 20.6% grave. Un 23.3% presentaron hematoma epidural, un 14.7% contusión cerebral y un 8.8% hemorragia subaracnoidea (HSA). En el 14.7% persisten secuelas neurológicas moderadas-severas. - No se encontraron anomalías en la exploración endocrinológica. La talla-SDS media fue -0,18±0,9 y el peso-SDS medio 0,1±1,2. En el estudio hormonal se objetivaron dos casos compatibles con insuficiencia suprarrenal secundaria. En cuatro niños los valores de IGFI estaban en el límite bajo de la normalidad. Estos pacientes se encuentran actualmente en seguimiento. No existe relación entre la presencia de alteración y una mayor gravedad del TCE.
Comentarios: - En esta serie es poco frecuente la alteración endocrinológica. La afectación del eje hipófiso-adrenal ha sido el único hallazgo valorable a diferencia de lo descrito en la literatura. - El TCE en los niños con afectación endocrina fue leve, de localización frontoparietal y con HSA.
P1/d2-3-132 PROLACTINOMA COMO CAUSA DE RETRASO PUBERAL. REVISIÓN DE 4 CASOS CON DISTINTA EVOLUCIÓN. A.M. Cabrejas Lalmolda, N. Martín Ruiz, A. De Arriba Muñoz, J.I. Perales Martinez, J.I. Labarta Aizpun, E. Mayayo Dehesa.
Hospital Infantil Miguel Servet, Zaragoza. Introducción: El prolactinoma es el tumor hipofisario más frecuente en la infancia. El retraso puberal y/o la no progresión es la forma más frecuente de debut en pediatría. Se presentan 4 casos de prolactinoma, 3 de ellos de evolución favorable y uno de evolución tórpida (ver tabla).
Caso 1: Varón de 89 años. Inicio de pubertad a los 11 años, con detención posterior. Velocidad de crecimiento 2’1cm/año. Estudio hormonal (12 años): prolactina (PRL):1024,3 ng/mL, test GRF, ITT, LHRH: disminuidos. RM: lesión hipofisaria de 18x18x16mm. Tratamiento: Cabergolina 0’5 mg/sem, RM control normal.
Actualmente cabergolina 0’25 mg/semana. Desarrollo puberal normal.
Caso 2: Mujer de 1210 años con obesidad y pubarquia II-III.A los 146 años cefalea y visión borrosa, realizándose neuroimagen: adenoma hipofisario 25’7x28’9x15’4mm., con extensión a región supraselar. PRL: 3276 ng/mL. Inicia cabergolina 0’5 mg/sem debiéndose aumentar a 4 mg/sem, manteniendo PRL de 750 ng/mL y mala tolerancia al tratamiento.
Cirugía transesfenoidal tras 9 meses de tratamiento, con extirpación incompleta por extensión tumoral hacia seno cavernoso derecho. Presenta déficit secundario de LH, FSH y GH. PRL>150 ng/mL, a pesar de cabergolina 4 mg/semana. Pendiente de radioterapia.
Caso 3: Mujer de 11 años con talla baja. Exploración: Tanner II
Caso 4: Mujer de 14 años. Madre: hipotiroidismo e hiperprolactinemia. Exploración: Obesidad generalizada. Estadio puberal: Tanner III. No menarquia. PRL: 379,64. RMN: adenoma hipofisario 10x13 mm. con pequeño foco hemorrágico. Tratamiento: Cabergolina 0’5 mg/sem. Aumento progresivo hasta 1,25 mg/sem. Menarquia a los 153 y pubertad completa.
Comentarios: El retraso en el inicio de la pubertad y/o la no progresión de la misma, obliga a descartar un prolactinoma. Nuestra experiencia indica que no todos los casos responden al tratamiento médico.
P1/d2-3-133 SÍNDROME POLIGLANDULAR AUTOINMUNE. DIFERENTES FORMAS DE PRESENTACIÓN. M. Royo Gómez, M.J. Olmos Jiménez, B. Huidobro Fernández, R. Rodríguez Caro, A. Rodríguez Sánchez de la Blanca, B. Roldán Martín.
Hospital General Universitario Gregorio Marañón, Madrid.
Introducción: El síndrome poliglandular autoinmune tipo I (SPA I) se define por la presencia de al menos dos de las siguientes entidades: candidiasis mucocutánea crónica, hipoparatiroidismo e insuficiencia suprarrenal, asociadas o no a otros defectos autoinmunes o endocrinos. Es una enfermedad de herencia autosómica recesiva cuyas manifestaciones suelen aparecer en la primera década y a lo largo de la vida de los pacientes. El gen afecto es denominado AIRE (autoinmune regulator) y las mutaciones en el gen son las responsables del desarrollo de la enfermedad.
Presentamos dos casos de SPA I:
Caso 1: Niña de 9 años y 8 meses, acude a urgencias por crisis convulsiva, detectándose hipocalcemia e hiperfosfatemia con niveles de PTH < 3 pg/dl. Fue diagnosticada de hipoparatiroidimo y tratada con calcitriol y calcio vía oral. Antecedentes personales: intervenida a los 2 años por ptosis palpebral congénita derecha, alopecia areata desde los 6 años y candidiasis oral recurrente. Desde los 7 años seguía tratamiento con corticoides por sospecha de lupus eritematoso sistémico en su país de origen. Fue diagnosticada de SPA tipo I y el estudio genético molecular del gen AIRE mostró la mutación p.Arg110GlysX37 (no descrita previamente).
Caso 2: Niño de 3 años derivado desde consultas de hematología para valoración de hiperpigmentación cutánea. Antecedentes personales: trombopenia secundaria a hipoplasia medular diagnosticada a los 17 meses y 2 episodios previos de decaimiento y deshidratación secundaria a emesis con hipoglucemia e hiponatremia. En estudio analítico se detectaron niveles elevados de ACTH así como hiperfosfatemia e hipocalcemia con valores de PTH < 3 pg/ml. Se determinó cortisol basal y tras estímulo con ACTH con resultados de 6.8 mcg/dl y de 7.38 mcg/dl respectivamente, compatible con enfermedad de Addison. Además asociaba candidiasis oral y manchas discrómicas. El estudio del gen AIRE resultó negativo.
Conclusiones: La aparición de alteraciones autoinmunes asociadas en un mismo paciente debe hacernos pensar en el diagnóstico de síndromes poliglandulares autoinmunes, sobre todo si se asocian a alteraciones funcionales de dos o más glándulas endocrinas. Es necesario el seguimiento a largo plazo de estos pacientes ya que ciertos componentes endocrinos pueden no aparecer hasta edades avanzadas.
P1/d2-3-134 SINDROME DE MARFÁN NEONATAL. A PROPÓSITO DE UN CASO. A.D. Alcalde De Alvaré, M. M. Hawkins Solís (1), J. Yebra Yebra (1), B. Pérez-Seoane Cuenca (1), T. Raga Poveda (1), Sixto García Miñaur (2).
(1) Hospital Infanta Sofía, (2) Hospital Universitario La Paz. Madrid.
Presentamos el caso de un neonato que ingresa al nacer por rasgos dismórficos: piel redundante y laxa, lipodistrofias en miembros inferiores, pectum excavatum, apófisis xifoidea prominente, cabello de implantación baja, hendiduras palpebrales antimongoloides, microretrognatia, labio superior fino, paladar ojival, aracnodactilia, desviación cubital de los dedos de las manos, pies valgos, limitación para la extensión articular, extremidades largas ( > 2SDS para la edad), llanto ronco, inexpresividad facial con escasa apertura ocular. Se aprecia soplo sistólico I/VI. Amniocentesis durante la gestación (cariotipo 46 XX) por sospecha de espina bífida oculta y ventrículomegalia.
En ecografía cerebral se evidencia megacisterna magna y tras valoración cardiológica se diagnóstica de CIA tipo Ostium Secundum, e insuficiencias aórtica y mitral leves, con válvula mitral displásica. Es seguido en consultas externas, donde tras valoración por el servicio de genética se realiza estudio molecular del gen FB1. En el estudio se detecta la presencia de la mutación c.3203G (p.CYs 1068Tyr) en heterocigosis en el exón 25 de dicho gen, confirmándose el diagnóstico de síndrome de Marfan neonatal. Se realiza RNM craneal donde se evidencia ventriculomegalia e hipodesarrollo del cerebelo. Precisa nutrición enteral continua domiciliaria a partir de los 10 meses de edad. Presenta empeoramiento progresivo de la cardiopatía con aparición de una insuficiencia mitral moderada-severa y una importante dilatación de la aurícula izquierda, con dilatación de la raíz aórtica e hipertensión pulmonar que desemboca en la aparición de una insuficiencia cardíaca severa refractaria al tratamiento farmacológico.
Se realiza estudio genético a todos los familiares de primer grado, que demuestra una mutación de novo. Desde los 4 meses de edad presenta varias infecciones respiratorias que precisan ingreso hospitalario, dos de ellos con asistencia respiratoria. Fallece durante el último ingreso, a los catorce meses de edad, por insuficiencia cardíaca, en el contexto de gastroenteritis aguda por Rotavirus.
Comentario: El síndrome de Marfan es una entidad bien conocida, pero cuya presentación neonatal tiene una escasa incidencia. La esperanza de vida suele ser de un año, con una mortalidad debida, sobre todo, a insuficiencia cardíaca o dilatación de la raíz aórtica.
P1/d2-3-135 SÍNDROME DE LEOPARD: CARACTERIZACIÓN FENOTÍPICA Y GENOTÍPICA. C.E. Heredia Ramírez (1), L. Castro-Feijóo (1), E. Balboa (2), J. Barreiro Conde (1), P. Cabanas Rodríguez (1),
(1) Unidad de Endocrinología Pediátrica, Crecimiento y Adolescencia. Hospital Clínico Universitario y Universidad de Santiago de Compostela (2) Fundación Pública Galega de Medicina Xenómica, CIBERER. (3) Cardiología Pediátrica. Departamento de Pediatría Hospital Clínico Universitario de Santiago de Compostela.
El síndrome de Leopard (SL) es un trastorno autosómico dominante que se superpone con el síndrome de Noonan (SN) con un espectro restringido de mutaciones en los genes PTPN11, BRAF y RAF1. Las características clínicas de presentación incluyen lentigos múltiples y defectos cardíacos.
Metodología: Se estudian 3 pacientes con sospecha clínica de SL, efectuando la caracterización fenotípica y el estudio molecular de los mismos, mediante secuenciación cíclica tras amplificación por PCR de toda la región codificante y los bordes intrón/exón del gen PTPN11.
Resultados: Son 2 pacientes mujeres (2,5 años y 7,3 años) y un varón (13,4 años). Fenotípicamente las dos niñas tienen facies típicas de SN; en todos se observa pabellones auriculares de implantación baja y rotación posterior, implantación posterior baja del cabello, paladar ojival y cuello corto; en dos pacientes se aprecia pterigium colli e hipertelorismo. Cardiológicamente las dos pacientes presentan cardiomiopatía hipertrófica (CMH) y dos pacientes alteraciones en el ECG (hemibloqueo anterior izquierdo y desviación del eje a la izquierda). En todos los pacientes se observa la presencia de múltiples lentigos; una paciente con una hipoacusia conductiva actualmente en resolución, pero ninguno con hipoacusia neurosensorial. Los tres pacientes tienen talla baja inferior a -2 SDS, dos con VC normales para la edad. Una paciente tiene un retraso mental leve con CI 75-86 y otra retraso del desarrollo psicomotor.
Análisis molecular: Se han encontrado tres mutaciones diferentes en el gen PTPN11 (G464A, T468M y Y279C), asociadas previamente al SL. El estudio familiar destaca que la madre del paciente varón era portadora de la misma mutación y una paciente tiene una mutación de novo.
Conclusiones: El SL es una enfermedad infrecuente, caracterizándose por la presencia de múltiples lentigos, anormalidades en el ECG, hipertelorismo ocular, CMH, retardo en el crecimiento y grado variable de discapacidad neurológica. Es de destacar que los pacientes deben llevar un seguimiento cercano por parte del pediatra realizándose una valoración multidisciplinaria, además de efectuarse el estudio genético familiar para confirmar el diagnóstico y dar un consejo genético.
P1/d2-3-136 PSEUDOHIPOPARATIROIDISMO E HIPERTENSIÓN ARTERIAL. ¿ENTIDADES ASOCIADAS? V. Recio Pascual, M.L. Fernández Pérez, L. Regueras Santos, A. Díaz Moro.
Complejo Asistencial Universitario de León, León.
Introducción: El pseudohipoparatiroidismo (PHP) comprende un grupo heterogéneo de entidades clínicas causadas por un defecto en la acción periférica de la paratohormona (PTH). Se caracteriza por hipocalcemia, hiperfosfatemia y niveles altos de PTH en sangre. La asociación entre hipertensión arterial (HTA) y PHP es conocida, existiendo sin embargo múltiples hipótesis sobre su patogenia, todavía no aclarada. Alteraciones en el sistema renina-angiotensina, en el sistema nervioso simpático o la propia PTH en exceso podrían ser los responsables.
Caso clínico: Varón de 3 años y 11 meses con episodios repetidos de espasmos carpo-pedales. No antecedentes personales de interés, ni enfermedades familiares conocidas salvo HTA en rama materna.
A la exploración presenta reflejos osteotendinosos exaltados, retracción en ambos pies y signos de Chvostek y Trousseau positivos, siendo el resto normal. Fenotipo normal con peso y talla en percentil 90 para su edad.
Ante sospecha de crisis de tetania se inicia estudio. Analítica con calcio 6,1 mg/dl, calcio iónico 2,5 mg/dl, fósforo 8,79 mg/dl, potasio, magnesio, albúmina y perfil lipídico normales, Vit 1,25 (OH)2D normal, fosfatasa alcalina 1.073 UI/l, osteocalcina 22,4 ng/ml y PTH 627 pcg/ml. En orina de 24 horas presenta hipocalciuria y reabsorción tubular de fosfato aumentada. Serie ósea sin alteraciones. Ante diagnóstico de PHP se inicia tratamiento con carbonato cálcico y calcitriol. Se realiza estudio hormonal hipotálamo-hipofisario resultando valores normales, salvo detección de hipotiroidismo, por lo que se pauta tratamiento con tiroxina. Precisa múltiples ingresos y continuos ajustes de las dosis por mal cumplimiento terapéutico. A los 10 años ingresa en nuestro hospital por episodio de crisis hipertensiva, constatándose cifras de tensión arterial mayores al percentil 95 para su edad, iniciándose tratamiento con un IECA sin conseguirse un adecuado control.
Conclusiones: En el PHP, además de un estudio inicial de múltiples hormonas por su posible resistencia asociada, no debemos olvidar un control riguroso de la tensión arterial, por el riesgo no menospreciado de desarrollar HTA. A pesar de la dificultad de alcanzar un buen control terapéutico en el PHP, si se consigue podría llegar a mejorar las cifras de tensión arterial por sí mismo.
P1/d2-3-137 SÍNDROME DE MORSIER DE PRESENTACIÓN NEONATAL. I. Costa Alcácer (1), M.J.López García (2), M.A. Moreno Ruíz (1), E. Ruíz González (1), E. Lucas Sáez (1), J. Vizuete (3).
(1) Servicio de Pediatría. Hospital de Manises, (2) Unidad de Endocrinología Pediátrica. Hospital Clínico Universitario de Valencia, (3) Servicio de Radiología. Hospital de Manises, Valencia.
Introducción: El síndrome de Morsier o displasia septo-óptica (DSO) es un trastorno clínicamente heterogéneo, caracterizado por alteraciones en la línea media, atrofia óptica e insuficiencia hipofisaria. Se describe un caso de diagnóstico neonatal y se resalta la importancia de su sospecha ante manifestaciones precoces. Caso clínico: Antecedentes: RNAT (41 +1)/PEG (PN= 2725); (L= 48). Cesárea. Apgar 8/10. Ingresa a las 34 h de vida por hipoglucemia sintomática que cede con glucosa IV. En los 3-4 días siguientes, glucemias límite, con normalización posterior. Estudio del equilibrio ácido-base, electrolitos y valoración hormonal (cortisol e insulina) normales. Ecografía suprarrenal normal. Reingresa al mes de vida por presentar irritabilidad y rechazo alimentario. Exploración: P= 3400 (+675 gr/40 días); constantes normales, bien hidratada. No fijación de la mirada y nistagmus giratorio. Hipotonía troncular. Resto normal.
Exploraciones complementarias: Na: 153 mEq/L; K= 5.4 mEq/L; Osmolaridad plasmática: 293 mOsm/kg. Volumen de diuresis (8 ml/kg/día). Densidad orina < 1005. Osmolaridad urinaria: 131 mOsm/L. Se completa estudio hormonal, con el hallazgo de hipotiroidismo central (ver tabla). La RNM muestra agenesia del septum pellucidum con hipoplasia del quiasma y nervios ópticos. No se identifica el brillo de la neurohipófisis. La adenohipófisis ocupa la silla turca. Diagnóstico: síndrome de Morsier.
Comentarios: 1) El reconocimiento temprano de la enfermedad (como es nuestro caso) previene de la muerte súbita en la DSO. 2) Destacar que el déficit hormonal más llamativo en nuestra paciente, (ADH), en la bibliografía se refiere como el menos frecuente asociado. 3) Recordar la importancia de las hipoglucemias neonatales como primera manifestación de un déficit hormonal. 4) Sugerir que ante el hallazgo de hipoplasia del nervio óptico deben realizarse estudios para descartar DSO y otras posibles alteraciones endocrinas para lograr un mejor manejo del paciente.
P1/d2-3-138 DIAGNÓSTICO GENÉTICO DE PSEUDOHIPOPARATIROIDISMO TIPO IB SIN MANIFESTACIONES CLÍNICAS. N. Gilabert Martínez (1), M.D. Zapico Álvarez-Cascos (1), G.Pérez de Nanclares(2), L. Ruiz Pérez (1), B. Castillo Gómez (1), M.D.Cañas Redondo (1).
(1) Unidad de Endocrinología Infantil. Hospital General de Alicante; (2) Grupo de Investigación de Endocrinología y Diabetes. Hospital de Cruces, Barakaldo.
Introducción: El pseudohipoparatiroidismo (PHP) está caracterizado por hipocalcemia e hiperfosfatemia debidas a resistencia a hormona paratiroidea (PTH). La causa genética de varios de los diferentes tipos de PHP es un trastorno en la proteína Gsα. Debido a las peculiaridades de esta herencia genética hemos decidido presentar este caso.
Caso clínico: Niño de 14 años remitido para estudio de hipocalcemia hallada de forma casual en un cuadro de rabdomiolisis aguda (Calcio 6,8 mg/dl, fosfato 7,1 mg/dl). Se inició tratamiento con vitamina D y calcio oral. Exploración física: Peso 82,8 kg (>p97). Talla 1,71 cm (p50-75). Fenotipo normal. Inteligencia acorde a su edad. Desarrollo puberal completo. No anomalías óseas.
Exploraciones complementarias: Calcio 9,4 mg/dl (en tratamiento), fosfato 6,3 mg/dl, PTH 442 pg/ml, 25-OH-vit D 17.1 ng/dl (en tratamiento). Función tiroidea normal.
Ante la sospecha de PHP se solicita estudio genético que confirma la presencia de pérdida en la metilación completa en los exones NESPas, XLαs y A/B. Esta alteración se asocia a PHP tipo Ib esporádico ya que el estudio genético de la madre es normal. El estudio de la deleción en NESPas y NESP55, asociado previamente a esta alteración de la metilación resulta negativo, asimismo se descarta la isodisomía.
El calcio se ha mantenido normal en sucesivos controles, continuándose el mismo tratamiento y encontrándose el paciente asintomático en todo momento.
Conclusiones. La mayoría de los casos de PHP-Ib esporádicos presentan alteraciones en la impronta del locus GNAS que codifica la proteína Gsα. El defecto más común es la pérdida de metilación en la DMR (differentialy methylated regions) del exón A/B en el alelo heredado de la madre, lo que lleva a una expresión bialélica del trascrito A/B. Se han descartado, en una amplia serie de casos de PHP-Ib esporádicos, deleciones que conllevan la pérdida completa de toda la DMR NESP55 mediante el análisis de polimorfismos en esta región. El PHP en la edad pediátrica es muy infrecuente pero debemos incluirlo siempre dentro del diagnóstico diferencial de cualquier hipocalcemia hallada de forma casual. El estudio genético resulta de gran utilidad tanto para confirmar el diagnóstico como para el consejo genético.
P1/d2-3-139 OSTEODISTROFIA HEREDITARIA DE ALBRIGHT Y DIABETES INSÍPIDA ¿ASOCIACIÓN O CASUALIDAD? M.J. Alcázar Villar (1), M.J. Rivero Martín (1), D. Martínez Sánchez (2), M. Sanz Fernández (1), C. Navarro Moreno (1), A. Barral Serrano (3).
(1) Servicio de Pediatría, Hospital Universitario de Fuenlabrada; (2) Servicio de Dermatología, Hospital Universitario de Fuenlabrada; (3) Servicio de Laboratorio Clínico, Hospital Universitario de Fuenlabrada, Madrid.
Introducción: El pseudohipoparatiroidismo (PHP) se caracteriza por resistencia a la hormona paratiroidea (PTH). Los pacientes con PHP tipo I pueden presentar otras resistencias hormonales y un fenotipo característico, conocido como osteodistrofia hereditaria de Albright (OHA), que asocia talla baja, obesidad, facies redonda y braquidactilia, además de calcificaciones cutáneas y discapacidad cognitiva. Los pacientes con PHP tipo II tienen como único hallazgo resistencia a la PTH.
Caso clínico: Primer hijo de padres no consanguíneos. Madre: anemia hemolítica, portadora de hepatitis C, hipotiroidismo en tratamiento con L-tiroxina y calcicosis cutáneas. Padre sano. Ecografía prenatal (36 semanas): macroglosia, huesos cortos e hidronefrosis grado III drcha. Cesárea a las 41.6 semanas, Apgar: 6/9. REA:III. Peso: 3.320 gr (-0.88DS), talla: 49 cm (-1.3DS), PC: 37 cm (0.95DS). Fenotipo: orejas de implantación límite, protrusión lingual, discreto pliegue nucal y pliegue simiesco bilateral. Ingreso en neonatos por hipoglucemia precoz y distrés respiratorio inmediato. 3er día de vida: TSH: 50.4 mcUI/ml, T4L:0.96 ng/ml, Ac. antitiroideos: 285 UI/ml. Ecografía y gammagrafía tiroidea normales. Cariotipo: 46 XY. Inicia tratamiento con levotiroxina. Evolución: Renograma diurético con patrón renal izquierdo obstructivo, intervenido a los 3 meses. Calcificaciones cutáneas a los 6.5 m (biopsia: osteoma). Metabolismo Ca/P: normal. PTH: 103 pg/ml.
Tratamiento: Calcio, vitamina D, calcitriol y dieta baja en fosfatos. Poliuria y polidipsia desde los 8 meses de vida con osmolaridad urinaria repetida de 1.000. Se realiza test de restricción hídrica y test de desmopresina compatible con diabetes insípida central parcial iniciando tratamiento con desmopresina oral y restricción hidrica. A los 13 meses: crisis tónico-clónica generalizada con Na capilar:118,3 mmol/l y calcio iónico normal. Precisa ajuste de dosis de desmopresina. IGF1:44 ng/ml (55 - 327). Estudio genético: mutación en el exón 7, consistente en c.565_568delGACT;p.Aspl89fs. RNM: sin alteraciones.
Comentarios: La diabetes insípida central no está descrita entre las resistencias hormonales asociadas a la OHA. El control del metabolismo del calcio disminuirá el riesgo de aparición de calcificaciones. Dificultad de tratamiento y riesgo de intoxicación acuosa en caso de administrar un exceso de líquidos junto con el tratamiento sustitutivo en pacientes de esta edad. Control evolutivo para la detección precoz de otros déficits hormonales.
P1/d2-3-140 RECIÉN NACIDO CON HIPOGLUCEMIA, ICTERICIA PROLONGADA E HIPOTONÍA. B. Fernández Valle (1), M.J. Ballester Herrera (1), L. Santillana Ferrer (2), M.D. Martínez Jiménez (2), E. Palomo Atance (1), E. Martin Campagne.
(1) Sección de Endocrinología pediátrica. Hospital General de Ciudad Real (2).Sección de Neonatología. HGCR, Ciudad Real.
Introducción: El hipopituitarismo congénito se define como la deficiencia de una o varias hormonas hipofisarias con expresión clínica variable. Puede estar causado por un traumatismo perinatal o alteraciones en el desarrollo del SNC asociado o no a mutaciones genéticas.
Caso clínico: Recién nacida a término, parto eutócico y somatometría normal que ingresa en neonatología por distrés respiratorio inmediato al nacimiento. En una ecografía prenatal (semana 21), describen ausencia de visualización de la porción anterior del cuerpo calloso, realizándose RMN fetal (semana 22), donde no se confirma dicha alteración. En la exploración física destaca hipotonía axial, ictericia precoz y fenotipo peculiar con hipertelorismo, raíz nasal ancha y narinas antevertidas. Presenta hipoglucemias recurrentes e hiperbilirrubinemia no iso- En la ecografía cerebral postnatal no se identifica el cuerpo calloso. La RMN cerebral muestra agenesia de la porción dorsal del mismo, con porción ventral hipoplásica. A su vez presenta un meningocele por defecto de la lámina cribosa y etmoides. El tallo hipofisario no se identifica en ninguna de las secuencias y se describe una silla turca poco excavada.
Los potenciales evocados visuales y el cariotipo resultaron normales. Se solicitó estudio de los genes relacionados con deficiencias hipofisarias múltiples, cuyo resultado está pendiente.
Se inició tratamiento con hidrocortisona, levotiroxina y posteriormente somatotropina.
Presenta, con 4 meses de vida adecuado desarrollo ponderoestatural y neurológico, así como resolución de la colestasis. Está pendiente de intervención neuroquirúrgica para extirpación del mielomeningocele.
Conclusiones: El diagnóstico del hipopituitarismo congénito se basa en la sospecha clínica en el periodo neonatal, caracterizándose por hipotonía y letargia con hipoglucemia, ictericia precoz y prolongada con/sin colestasis, hipogenitalismo en los varones más alteraciones de la línea media. A pesar de su baja incidencia (< 3 casos/millón/año), es obligado realizar estudio de la función hipotalamo-hipofisaria ante defectos en la línea media, para realizar un diagnóstico precoz y comenzar tratamiento sustitutivo temprano.
P1/d2-3-141 SÍNDROME DE MOEBIUS ASOCIADO A DEFICIENCIAS HIPOFISARIAS MÚLTIPLES. M.J. Ballester Herrera, E. Palomo Atance, E. Martín Campagne, L. Santillana Ferrer, M.J. Sánchez Fernández, P. Donado Palencia.
Hospital General Ciudad Real, Ciudad Real.
Introducción: El síndrome de Moebius es una alteración congénita poco frecuente caracterizada por parálisis del nervio facial, completa o parcial, unilateral o bilateral. Pueden afectarse otros pares craneales por aplasia o hipoplasia de los correspondientes núcleos encefálicos. Entre sus posibles causas están las alteraciones genéticas, la exposición prenatal a fármacos o la disrupción vascular.
Caso clínico: Niño de 14 años, derivado a consulta de Endocrinología pediátrica por hipotiroidismo central en tratamiento con levotiroxina. Entre los antecedentes personales destaca el diagnóstico de síndrome de Moebius en el periodo neonatal con parálisis facial bilateral, así como del III y VI par. En la exploración física se evidencian los rasgos fenotípicos característicos: facies inexpresiva (cara en máscara), estrabismo bilateral convergente, ptosis palpebral, micrognatia y paladar ojival. Así mismo se evidencia talla baja en -4,4 D.E, obesidad central (IMC de 34,5 kg/cm2; +3,4 DE), micropene y criptorquídia bilateral. Presenta retraso mental moderado. Se solicita ecografía abdominal en la que se evidencian ambos testículos en canal inguinal, edad ósea (13 años y 6 meses) y RMN cerebral, en la que no existen alteraciones hipofisarias y sólo destaca una megacisterna magna.
En las determinaciones hormonales se encuentran niveles bajos de TSH:0,3 mUI/ml; LH:0,4 mUI/ml; FSH: 1,46 m UI/ml; Testosterona: 0,1 ng/mL; IGF-I: 50 ng/ml; IGFBP-3: 2,25mcg/ml y GH:<0,05 ng/ml. Presenta niveles normales de ACTH, cortisol basal y prolactina. Para el estudio dinámico del eje somatotropo se realizan test de estímulo con clonidina e hipoglucemia insulínica con picos máximos de GH inferiores a 0,05 ng/ml.
Se inicia tratamiento con somatotropina a dosis de 1 mg/m2sc/día, continuando con levotiroxina. El paciente va a ser intervenido de criptorquidia bilateral, y tras la cirugía será valorado el eje hipotálamo-hipofisario-gonadal para considerar la inducción del desarrollo puberal.
Conclusiones: Se describe una forma de presentación poco frecuente del síndrome de Moebius, asociada a deficiencia múltiple de hormonas hipofisarias. Están descritos casos con hipogonadismo hipogonadotropo, sin embargo no se han publicado formas asociadas a las alteraciones hormonales múltiples recogidas en este paciente.
P1/d2-3-142 DIABETES INSÍPIDA CENTRAL ASOCIADA A DÉFICIT DE GH. C. Reig del Moral, L. García Blázquez, R. Alcedo Olea, B. Hernández Macho, C. de las Heras Díaz-Varela, A. García Rodríguez.
Hospital General de Segovia, Segovia.
Introducción: La diabetes insípida central producida por lesión de la neurohipófisis puede tener un origen genético o adquirido (tumoral, malformativo, inflamatorio, traumático...). Un 20 -50% de los casos son de origen idiopático. La presencia de alteraciones de adenohipófisis y neurohipófisis simultáneas resulta inusual sin evidencia de causa orgánica. Los tumores representan el 10-15% de los casos hallándose frecuentemente engrosamiento parcial o total del tallo hipofisario.
Caso Clínico: Niño de 7 años con progresivo sobrepeso y detención del crecimiento con pérdida de 2,8 DS desde los 4 años, junto con poliuria y polidipsia de 4 litros/día. Antecedentes: abuelo materno fallecido por astrocitoma presentaba polidipsia. Embarazo gemelar con pérdida fetal a las 10 semanas. Cesárea a las 35 semanas. Apgar 10/10. Somatometría neonatal normal. Presentó taquipnea transitoria, hipoglucemia leve e ictericia neonatales. Exploración física actual normal con peso en p85, talla en p14 e IMC en p98. Diuresis 24h: 3900ml (5.2ml/kg/h). Analítica basal: Na 139 mEq/l, Osm sangre/orina: 285/103 mOs/l , Creatinina 0,3 mg/dl. Hematimetria y bioquímica general normales. Tras deprivación hídrica durante 17 h: Osm sangre/orina: 302/490 mOs/l. Na 144 mE/l. Tras administración posterior de desmopresina: Osm sangre/orina: 301/697 mOs/l, IGF1 23ng/ml; IGFBP3 2,24 mg/L. Pico de GH: 2,9 ng/ml (glucagón-propanolol) y 1,2 ng/ml (hipoglucemia insulínica). Cortisol: 14,62 mcg/dl, prolactina 8,5 ng/ml. Test de TSH y LHRH normales. Autoinmunidad y marcadores tumorales negativos. Serie ósea sin lesiones líticas. Edad ósea: 7 años. RNM: hipoplasia de hipófisis y tallo con ausencia de señal de neurohipófisis. Genética: AVPR2 y HEX1 negativos. Diagnóstico: Déficit total de GH y parcial de ADH de origen central y etiología idiopática.
Evolución: Tras 2 años y medio de tratamiento con GH (25-30 mcg/kg/día) y desmopresina (180 mcg/día) presenta recuperación de talla y normalización de la diuresis con controles analíticos normales. No se han presentado otros déficit hipofisarios. Controles de RNM sin cambios.
Conclusiones: Se propone una etiología disgenética congénita en este caso que presenta hipoplasia de hipófisis y tallo con ausencia de señal neurohipofisaria aunque es conveniente realizar seguimiento estricto con controles de RNM para descartar origen tumoral.
P1/d2-3-143 FARMACOCINÉTICA DE LA METFORMINA EN NIÑAS CON PUBERTAD ADELANTADA Y BAJO PESO AL NACER PARA LA EDAD GESTACIONAL. D. Sánchez-Infantes Sánchez (1), M. Díaz (1,2), M.V. Marcos (2,4), A. López-Bermejo (3), F. de Zegher (5), L. Ibáñez (1,2).
(1) Endocrinología, Hospital Sant Joan de Déu, Universitat de Barcelona, (2) CIBER de Diabetes y Enfermedades Metabólicas Asociadas (CIBERDEM), (3) Endocrinología, Hospital Dr. Josep Trueta, Girona, (4) Endocrinología, Hospital de Terrassa, Terrassa, (5) Department of Woman & Child, Universidad de Lovaina, Bélgica.
Antecedentes: La metformina es una biguanida habitualmente utilizada en el tratamiento de la diabetes tipo 2. En niñas con pubertad adelantada rápidamente evolutiva y bajo peso al nacer, la metformina modula el tempo puberal y normaliza la edad de la menarquia. La farmacocinética de la metformina después de su administración oral sólo ha sido estudiada en un número reducido de adultos y adolescentes mayores de 12 años con diabetes tipo 2.
Objetivo: Determinar por primera vez en niñas menores de 10 años los parámetros farmacocinéticos de la metformina a lo largo de 12 horas después de la ingesta oral del fármaco.
Pacientes y Métodos: Se estudiaron 6 niñas [edad (promedio ± ES):
Resultados: La dosis promedio de metformina fue de 26.5 ± 2.4 mg/Kg; el AUC fue de 21mg.h/L; la Cmax fue de 3 mg/L y se alcanzó en 2,5 horas [tmax]. El t ½ fue de 4 horas y el aclaramiento de 20 L/h. La variabilidad interindividual fue mínima en las pacientes que recibieron la dosis de 850 mg.
Conclusión: En niñas no obesas de edad inferior a 10 años, la farmacocinética de la metformina es similar a la descrita en adultos y adolescentes con diabetes tipo 2. Una dosis diaria de 850 mg, considerada clínicamente efectiva en la modulación del tempo puberal, es segura, ya que determina una Cmax por debajo del rango tóxico, y es bien tolerada en pacientes de esta edad. |
| References |
 |
 |
 |
 |
 |
 Print
Print View / Download PDF
View / Download PDF  English version
English version Comments
Comments Add Favorites
Add Favorites