
| Rev Esp Endocrinol Pediatr 2011;2 Suppl(1):93-104 | Doi. 10.3266/RevEspEndocrinologPediatr.pre2011.May.55 |
| Crecimiento |
| Sent for review: 2 May. 2011 | Accepted: 3 May. 2011 | Published: 3 May. 2011 |
| Congreso SEEP |
| Correspondence: Congreso SEEP E-mail: seep@seep |
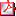 Tabla - P1d2-3-002_012_Estudio de Crecimiento andaluz Tabla - P1d2-3-002_012_Estudio de Crecimiento andaluz |
| Tabla - P1D2-3-003_012_Talla baja asociada a mutación |
| Tabla - P1d2-3-005_012_hipoplasia_hipofisaria |
| Tabla - P1d2-3-007_012_Diferencias auxológicas, clínicas y analíticas entre el déficit idiopático (DI) y el déficit orgánico (DO) de hormona de crecimiento (GH) |
| Tabla - P1d2-3-013_GH talla final |
| Tabla - P1d2-3-015_012_estudio_costes_GH |
| Tabla - P1d2-3-017_012_Evolución IGF1, IGFBP3 y cociente |
|
|
P1/d2-3-001 TALLA BAJA CON ALTERACIÓN DEL EJE GH-IGF1. NUEVA APORTACIÓN AL COMPLEJO SINDRÓMICO HIPER-IGD. L. Castro Feijóo (1), J.M. Couselo (2), L. Loidi (3), C. Quinteiro (3), P. Cabanas (1), C. Heredia (1), J. Barreiro (1),
(1) Unidad de Endocrinología Pediátrica, Crecimiento y Adolescencia. Hospital Clínico Universitario y Universidad de Santiago de Compostela, (2) Unidad de Oncohematología Pediátrica. Hospital Clínico Universitario de Santiago de Compostela, (3) Unidad de Medicina Molecular. Fundación Pública Galega de Medicina Xenómica. Santiago de Compostela, A Coruña.
La vía mevalonato es vital en múltiples procesos metabólicos. La disminución de actividad de la enzima mevalonato-quinasa (MK) se ha identificado como causa de la aciduria mevalónica y del síndrome hiperinmunoglobulinemia D y fiebre periódica (síndrome hiper-IgD). Investigaciones genéticas recientes han identificado en estos cuadros mutaciones en el gen MVK (12q24). El síndrome hiper-IgD, de herencia AR, se caracteriza por procesos febriles recurrentes asociados a IgD elevada. El nexo entre deficiencia de MK e inflamación no se conoce, pero parece estar relacionado con la estimulación del sistema inmune que explicaría los niveles elevados de IgD. En nuestro conocimiento, no existe descripción de alteraciones en el eje GH-IGF1 en este tipo de pacientes.
Caso clínico Niña remitida a los 8,8 años de edad para estudio de talla baja con sospecha de síndrome de Laron. Presenta fenotipo peculiar: frente amplia, micrognatia, clinodactilia y voz aguda. Talla: SDS – 4,54; TD: SDS-1,78; M1P1; MO retrasada. Antecedentes: anemia leve-moderada. Dolores articulares y/o cefalea ocasional. Episodios febriles recurrentes y rash urticariforme. Estudio del eje GH-IGF1: IGF1 e IGFBP3 reiteradamente disminuidos. GH: pico de respuesta 19 ng/mL, test de generación de IGF1 con pobre respuesta. Estudio de genes RGH e IGF1: sin alteraciones significativas. Ante la sospecha de etiopatogenia hematológica se efectúa estudio: aspirado de médula ósea, normal. IgD sérica elevada. Gen TNFRSF1A: No mutaciones. Gen MVK: Pendiente secuenciación completa.
Diagnóstico/Evolución Síndrome hiper-IgD. Sigue tratamiento con Anakinra normalizándose los valores de IgD, con desa-
Conclusiones 1. Se describe por primera vez, en nuestro conocimiento, la presencia de talla baja con alteración del eje GH-IGF1 en el síndrome hiper-IgD. 2. Consideramos que la afectación de la vía mevalonato-quinasa ha ocasionado un cuadro inmunológico e inflamatorio crónico que ha repercutido en el eje GH-IGF1 condicionando una talla baja extrema con signos bioquímicos de resistencia a la GH. 3. El tratamiento con Anakinra ha normalizado la afectación inmunológica, hematológica y endocrina de la paciente repercutiendo positivamente en el crecimiento. 4. Se demuestra la importancia del estudio minucioso del paciente con talla baja.
P1/d2-3-002 ESTUDIO LONGITUDINAL DE CRECIMIENTO EN ANDALUCÍA (0-3 AÑOS). J.P. López Siguero(3), J.M. Fernández García (1), J.D. Luna Castillo (2) J.M. Jiménez Hinojosa (3), C. Ruiz Cosano (1), A. Jurado Ortíz (3).
(1) Hospital Clínico, Granada, (2) Facultad de Medicina, Granada, (3) Hospital Materno-Infantil, Málaga.
Objetivo: Obtener los datos de normalidad de longitud, peso y perímetro cefálico de niños andaluces entre los 0 y 36 meses de edad.
Diseño: Se tomó una muestra de 500 recién nacidos de dos hospitales públicos andaluces (Materno-Infantil de Málaga y Clínico de Granada) que cubren el 20% de la población de Andalucía. Se midieron las variables: peso, longitud y perímetro cefálico a los 0, 3, 6, 9, 12, 18, 24, 30 y 36 meses de vida.
Metodología: Primer ajuste descriptivo de las variables, tiempo a tiempo, con objeto de detectar datos extremos. Se calcularon los percentiles: 1, 3, 5, 10, 25, 50, 75, 90, 95, 97 y 99. Para el ajuste de las curvas se empleó el método de GAMLSS ajustando con la distribución más flexible. Para cada variable se probó la distribución más ajustada, empleando como criterio el de información de Akaike y como medidas de calidad el ajuste de los modelos. Se construyeron las curvas con intervalo semestral de 0 a 36 meses.
Resultados: La muestra final fue de 458 niños con 135 (29,5%) que llegaron a los 36 meses. Los resultados se presentan en cm y Kg, como media (DS) en la siguiente tabla. Estos resultados no difieren significativamente de los obtenidos del estudio longitudinal español de 2010.
Conclusiones: 1. Se presentan los datos de longitud, peso y perímetro cefálico del primer estudio de crecimiento longitudinal andaluz entre 0 y 36 meses. 2. Estos datos no difieren de los del estudio longitudinal español de 2010.
P1/D2-3-003 TALLA BAJA ASOCIADA A MUTACIÓN EN HETEROCIGOSIS DEL RECEPTOR DE HORMONA DE CRECIMIENTO. F. Hermoso López, P. Bahíllo Curieses, P. García Gutiérrez, O. Zurita Muñoz, S. de Frutos, A. Campos Barros.
Hospital Clínico Universitario Valladolid. Instituto de Genética Médica y Molecular, IdiPaz, Hospital Universitario La Paz y CIBER de enfermedades raras (U.753).
Introducción: La talla baja en niños sin déficit de hormona de crecimiento (GH) puede ser debida a varias etiologías, y una vez descartadas alteraciones metabólicas-nutricionales, es importante la valoración del eje GH-IGF-1. Caso clínico: Niña de 3 años y 3 meses de origen búlgaro con hipocrecimiento armónico. Talla diana 153,5 cm. No consanguinidad. Somatometría normal al nacimiento. Exploración física: Peso 9.6 kg (-3.08 DS), talla 86.5 cm (-2.94 DS), IMC 12.83 kg/m2 (-2.44 DS), Perímetro cefálico: 46.5 cm (A (CGT>CAT p.R229H) en el exón 7 de GHR en heterocigosis. Se inicia tratamiento con rhIGF-1 a los 8 a 3 m (dosis inicial de 40 µg/kg/12h e incremento a 120 µg/kg en 30 días), con buena tolerancia y ausencia de efectos adversos. Conclusiones: Un porcentaje importante de niños catalogados de talla baja idiopática, presentan déficit primario de IGF-1. Entre las diferentes causas de déficit primario de IGF-1 se encuentra la insensibilidad extracelular al receptor de GH. En nuestra paciente se encontró una mutación de cambio de sentido c.686G>A (CGT>CAT p.R229H) en el exón 7 en heterocigosis descrita por primera vez por Goddard en 1995.
P1/d2-3-004 DISCONDROSTEOSIS DE LÉRI-WEILL CON ESCASA EXPRESIVIDAD CLÍNICA. S. de Murcia Lemauviel, C. Buhedo Gordillo, M. Romero Gómez, R. Miralles Chinchilla, C. Gleser, J. Frontela Losa.
Hospital de Torrevieja, Alicante.
Niño de 12.5 años remitido por talla inferior al p3. Inicio puberal a los 11.5 años con velocidad de crecimiento de 5 cm/año (- 2.9 DE). Perinatal normal (peso 3.150 g y talla 48 cm, a término, Apgar 9/10).
Exploración: Peso 31 Kg (- 1.6 DE), talla: 136.6 cm (- 2.4 DE), IMC 16.6 (p25), aparentemente proporcionado, envergadura: 135 cm (- 2.6 DE), envergadura/talla = 98.3%, talla sentada/talla = 52%, rodillas y tobillos con leve protrusión medial, no hipertrofia muscular. Tanner III. Se objetiva una disminución preferente de los segmentos corporales medios (antebrazo - 3.4 DE y pierna - 3 DE), sin deformidades óseas evidentes. Puntuación de 3 en el score de Rappold (probabilidad baja de alteración del gen SHOX). Su pronóstico de talla es de 162 cm con talla diana de 166 cm. El padre mide 168 cm, fenotipo armónico. La madre mide 151.5 cm, (- 2.09 DE), aparentemente proporcionada, sin deformidades óseas evidentes, envergadura 143 (- 3.05 DE), cociente SS/SI: 1.2 (elevado), envergadura/talla = 94.3%, talla sentada/talla = 55.1%), leve hipertrofia gemelar y protrusión femoral y tibial internas. Puntuación de 9 según Rappold (probabilidad media).
Dada la puntuación materna, se solicita una radiografía de antebrazos del niño y de la madre siendo compatibles con deformidad de Madelung leve en la actualidad. Se detecta una “delección del gen SHOX en la región pseudoautosómica del cromosoma X materno y del paciente, englobando también a la región PAR distal (PAR0cM, DXY233)”. Actualmente el niño se encuentra en tratamiento con GH con buena respuesta.
Discusión: La haploinsuficiencia del gen SHOX tiene una expresividad clínica muy variable pudiendo presentar incluso una talla normal. Las deformidades esqueléticas con frecuencia son leves pero progresivas, y difíciles de detectar en la infancia.
Dada su implicación terapéutica, consideramos que una puntuación baja en el score de Rappold no debería ser un criterio excluyente para realizar el estudio genético, debiendo tener en cuenta todas las características del paciente. Recordamos la importancia de una auxología exhaustiva, incluida la medición de los segmentos corporales, y de la búsqueda de anomalías óseas radiológicas leves en los pacientes con talla baja idiopática.
P1/d2-3-005 HIPOPLASIA HIPOFISARIA: NUESTRA CASÍSTICA. B. Jiménez Crespo, E. Gómez Santos, M.T. Ferrer Castillo, M.M. Román del Río, C. Hermoso Torregosa.
Hospital Juan Ramon Jiménez, Huelva.
Introducción: El hipopituitarismo congénito por hipoplasia de la hipófisis anterior y/o tallo hipofisario es un trastorno raro. La clínica puede estar presente al nacimiento en formas severas (hipoglucemia, ictericia prolongada, micropene…) o pasar desapercibida hasta etapas más avanzadas (crecimiento errático, retraso puberal…) La etiología puede ser genética o asociada a traumas o noxas perinatales. Han sido detectadas alteraciones en 5 genes (PIT1, PROP1, LHX3, LHX4 y HESX1).
Material y Métodos: Se revisaron retrospectivamente historias clínicas de 4 pacientes diagnosticados de talla baja secundaria a hipoplasia hipofisaria en los últimos 5 años, y se analizaron diversas variables: motivo consulta, antecedentes perinatales, edad al diagnóstico, respuesta test estímulo, exploración, hallazgos en neuroimagen y respuesta al tratamiento.
Resultados: Conclusiones: -En cualquier paciente con talla baja armónica extrema >-3DS sin enfermedad asociada, debería investigarse posible déficit de GH. -Ante un déficit severo, la RMN selar es obligada. -Los antecedentes familiares de talla baja extrema ponen en alerta de un posible déficit de GH genético. -En un déficit hormonal por hipoplasia hipofisaria, es preciso seguimiento del resto de ejes hormonales porque la expresión de la deficiencia puede ser progresiva en el tiempo. P1/d2-3-006 DESCRIPCIÓN DE LAS CARACTERÍSTICAS AUXOLÓGICAS Y CLÍNICAS DE UNA POBLACIÓN DE NIÑOS DIAGNOSTICADOS DE DÉFICIT DE HORMONA DE CRECIMIENTO (GH). A. Aguayo Calcena, M. Urrutia Adán, A. Rodríguez Estévez, G. Grau Bolado, I. Rica Etxebarria, A. Vela Desojo.
Hospital de Cruces, Barakaldo (Vizcaya).
El diagnóstico de déficit de GH es uno de los retos en la consulta de Endocrinología pediátrica.
Objetivo: Describir las características de una población de niños diagnosticados de déficit de GH.
Población y Metodología: Estudio retrospectivo de pacientes diagnosticados de déficit de GH entre los años 1999 y 2009. En ausencia de otra patología responsable del hipocrecimiento, con criterios auxológicos establecidos y con la presencia de dos respuestas < 10 ng/ml de GH tras estímulo. Definimos talla baja familiar si la talla paterna y/o materna es ≤ 165 cm y 150 cm respectivamente.
Resultados: Se han revisado 92 historias de niños. El 69% son varones con edad media al diagnóstico de 10,2 años (r: 1,33-15,38), no habiendo diferencias entre ambos sexos. El 71% son prepuberales. En el 96% la presentación en el parto es cefálica y en el 4% de nalgas. Presentan una longitud al nacimiento de -0,63 ± 1,6 SDS de media y un peso de -0,54 ± 1,17 SDS de media. El 13% de los pacientes tienen un peso < 2 SDS y el 10% una longitud < 2 SDS. La mayoría eran déficit idiopáticos (DI) (73%) y 27% déficit orgánicos (DO). El 40% tienen antecedentes de talla baja familiar (TBF) sin haber diferencias entre sexos. La TBF está presente en el 87% de los DI frente al 13% de los DO. La talla (SDS) al diagnóstico de toda la población era de (-2,81 ± 1), menor en niñas (-3,32 ± 1,03) de forma significativa (p<0,01). Comparamos la talla (SDS) al diagnóstico con la talla diana (SDS): -1,34 ± 1,1 sin diferencias entre ambos sexos. El IMC al diagnóstico era de -0,06 ± 1,25 y sólo el 5% de los pacientes eran obesos. No había diferencias entre ambos sexos.
Conclusiones: 1. La presentación de nalgas es poco frecuente. 2. La mayoría de los pacientes tiene una talla y/o peso adecuado al nacimiento. 3. En la mayoría, el déficit es idiopático. 4. Un porcentaje importante tiene TBF. 5. La presencia de TBF es más frecuente en el DI. 6. La talla de las niñas es menor antes del tratamiento. P1/d2-3-007 DIFERENCIAS AUXOLÓGICAS, CLÍNICAS Y ANALÍTICAS ENTRE EL DÉFICIT IDIOPÁTICO (DI) Y EL DÉFICIT ORGÁNICO (DO) DE HORMONA DE CRECIMIENTO (GH). A. Aguayo Calcena, M. Urrutia Adán, A. Rodríguez Estévez, G. Grau Bolado, I. Rica Etxebarria, A. Vela Desojo.
Hospital de Cruces, Barakaldo (Vizcaya).
El déficit de GH puede ser DI o DO. Además de la causa que provoca el DO puede haber otros factores que los diferencien e influyan en la respuesta final al tratamiento.
Objetivo: Comparar parámetros clínicos, analíticos y auxológicos entre ambos déficit y analizar si alguno de ellos influye en la respuesta al tratamiento.
Pacientes y Metodología: Estudio retrospectivo de pacientes diagnosticados de déficit de GH (criterios auxológicos establecidos, ausencia de otra patología responsable de hipocrecimiento, respuesta de GH < 10 ng/ml a dos estímulos diferentes). Valoramos respuesta máxima de GH a uno de los estímulos (GH-max), diferencia entre la edad cronológica y edad ósea (EC-EO). Valoramos la talla (SDS) al diagnóstico (TD). Comparamos la talla final (SDS) con su talla diana (SDS) mediante la variable DIF-T. Además valoramos la respuesta al tratamiento como positiva si alcanzaron o superaron su talla diana.
Resultados: Se han revisado 92 historias de niños en consulta entre los años 1999 y 2009 diagnosticados de déficit de GH y que hayan finalizado su tratamiento. El 32% son niñas, la mayoría prepuberales (71%). Las causas de DO eran congénitas/estructurales en 41% y el resto secundarias a tumores. El 40% presentan talla baja familiar, siendo más frecuente en los DI. La edad al diagnóstico (años) era de 10,2 ± 3,1 sin diferencias entre ambos grupos. Responden al tratamiento el 67%. Los niños respondedores, presentaban una diferencia menor con respecto a su talla diana al inicio del tratamiento. De los pacientes con TBF el 84% responden.
Conclusiones: 1.- No hay diferencias en la edad al diagnóstico en ambos déficit. 2.- La talla al diagnóstico es menor en los DI. 3.- La GH-máx es mayor en el DI. 4.- Los niños que responden al tratamiento, tienen una diferencia menor con su talla diana al inicio. 5.- Los pacientes con TBF responden mejor al tratamiento.
P1/d2-3-008 SÍNDROME DE LARON Y ALLGROVE EN UN MISMO PACIENTE. S. Marín del Barrio(1), P. Casano-Sancho (1), N. Villarreal Peña (1), G. Sebastiani (1), B. Pérez Dueñas (2), L. Ibáñez Toda (1).
(1) Servicio de Endocrinología. (2) Servicio de Neurología. Hospital Sant Joan de Déu, Esplugues, Barcelona.
Introducción: El síndrome de Laron y el síndrome de Allgrove son entidades muy poco prevalentes (1-9/1.000.000 y <1/1.000.000, respectivamente). Presentamos el caso de un paciente con ambas patologías.
Caso clínico: Niño de 7 años, procedente de Pakistán, que consulta por talla baja. Fruto de tercer embarazo a término. Peso y longitud al nacer desconocidos. Hijo de padres consanguíneos. Talla paterna 179 cm, talla materna 156 cm. Dos hermanos estudiados por talla baja.
Exploración: Destaca talla 96 cm (-5.6 DE), peso 14.6 Kg (-2.9 DE), IMC 15.8 Kg/m2 (-0.3 DE), obesidad troncular, frente prominente, puente nasal deprimido, cabello escaso, malposición dental y retraso cognitivo.
Exploraciones complementarias: Edad ósea 6 años. IGF1 37 ng/ml. Test de estimulación de GH (clonidina): GH basal 3,7 ng/ml, pico GH: 32,7 ng/ml. Test de generación de IGF1: IGF1 basal 31 ng/ml, post-GH (5 dosis; 0,05 mg/Kg): IGF128 ng/ml. RM hipófisis: normal. Análisis del gen del receptor de GH: mutación del pseudoexón 6 en homocigosis (resistencia total a la GH, síndrome de Laron).
Evolución: A los 9 años inicia tratamiento con IGF-I recombinante (dosis: 0,04 mg/kg/12h hasta 0,07 mg/kg/12h). Mejora la velocidad de crecimiento con normalización de IGF-I sérica. No hipoglucemias graves, pero requiere intervención por hipertrofia adenoidea. A los 10,5 años, presenta debilidad, pérdida de peso, anorexia, hiperpigmentación de mucosa oral y de zonas extensoras de articulaciones. Exploraciones complementarias: Cortisol 3 µg/dl, ACTH basal 689 pg/ml. Test de ACTH: cortisol basal 3 µg/dl, post-ACTH 3 µg/dl. 17-OH-progesterona normal. Anticuerpos antiadrenales negativos. Con el diagnóstico de insuficiencia suprarrenal primaria, inicia hidrocortisona a 13 mg/m2/día con franca mejoría clínica. A los 11 años, refiere disfagia a sólidos, diagnosticándose de acalasia que se corrige quirúrgicamente, y alacrimia, objetivándose una queratitis filamentosa por sequedad ocular. El estudio de genética molecular muestra una mutación en homocigosis del gen AAS (p.Arg194X), confirmándose el síndrome de Allgrove o Triple A.
Comentarios: No hay descritos casos en la literatura de pacientes con ambos síndromes, en los que además, los genes mutados se encuentran distantes en el genoma. A pesar de su rareza, un minucioso seguimiento clínico y las exploraciones complementarias adecuadas permitieron sospechar y confirmar el diagnóstico.
P1/d2-3-009 ¿TALLA BAJA NO FILIADA O TALLA BAJA IDIOPÁTICA? A.C. Rodríguez Dehli (1), I. Riaño Galán (1), I. Llano Rivas (2), J. Ignacio Suárez Tomás (1), J. Fernández Toral (2).
(1) Hospital San Agustín. Avilés; (2 ) Hospital Universitario Central de Asturias. Oviedo, Asturias.
Introducción: La talla baja idiopática es un diagnóstico por exclusión, siendo un cajón de sastre de hipocrecimientos de causa no conocida.
Caso clínico: Niña de 6 años y 2 meses con talla baja. Creció canalizada a -3 DS de talla y -2 DS de peso, iniciándose tratamiento con rGH (0,5 mg/sc día) con 4 años y 5 meses, con regular respuesta (talla inicio tratamiento en -3,12 DS y al año -4,2 DS; IGF I inicio 102 ng/ml; IGFBP3 2,39 mcg/ml y al año 84 y 2,43).
Antecedentes familiares: Madre: mide 152,1 cm, menarquia a los 15 años, esclerótica azulada. Padre mide 172,1 cm. Rama materna: tallas bajas. Abuela materna problemas óseos, esclerótica azulada. Hermano de 7 años con luxación congénita de rodillas y sospecha de displasia ósea no confirmada inicialmente al nacimiento. No consanguinidad. Antecedentes personales: Gestación con diabetes gestacional, 37 semanas. Al nacer P= 2190 g, L= 44 cm, PC= 32 cm. Hipoglucemias límites. Inapetente. Valgo de talón y medio pie.
Exploración física: T= 99,2 cm (-3,81 DS), P= 13,150 kg (-2,43 DS); 82,5% P/T. IMC 13,21 kg/m2 (-1,42 DS), envergadura 97 cm (acorde con talla), mano 12 cm, 3er dedo 5 cm (p3 y 10). Limitación de la flexión 1ª falange de los 2º y 3er dedos manos bilateral, dedos pulgares anchos, filtrum largo, soplo sistólico en 4º epi, tórax asimétrico levemente en quilla, valgo de talón, resto dentro de límites normales. Pruebas complementarias: serie ósea con anomalías en húmeros, falanges, epífisis femoral proximal y raquis. El estudio del gen SLC 26 A2 (displasia múltiple epifisaria tipo 4) en ambos hermanos muestra la mutación R279 W en el exón 3 en homocigosis y en los padres en heterocigosis. Actualmente: talla 103.5 cm (-3.77 DS); P= 14,4 kg (-2.27 DS). VC ha sido de 6,57 cm/año (P73, +0,61 DS), IGF I 243 ng/ml, IGFBP3 3.59 mcg/ml.
Comentarios: Las displasias óseas pueden ser visibles desde el momento del nacimiento o pasar desapercibidas al manifestarse tardíamente. En estos casos es necesaria una exploración física más exhaustiva que junto con los estudios de genética molecular nos pueda permitir llegar a un diagnóstico.
P1/d2-3-010 EXPOSICIÓN PRENATAL AL MERCURIO Y PLOMO Y CRECIMIENTO EN LA COHORTE INMA ASTURIAS. A.C. Rodríguez Dehli (1), I. Riaño Galán (1), I. García (1),
(1) Hospital San Agustín. Avilés; (2) Universidad de Oviedo. Oviedo, Asturias.
Introducción: La exposición prenatal a metales pesados como el mercurio y/o plomo podría afectar al crecimiento pre y postnatal.
Objetivos: Evaluar los niveles de plomo (Pb) y mercurio (Hg) medidos en sangre de cordón en la cohorte INMA Asturias y su influencia en el peso al nacer y la prematuridad
Métodos: La muestra está constituida por los 482 niños de la cohorte prospectiva de Asturias, dentro del proyecto multicéntrico INMA. Los niveles de Pb y Hg se analizaron en 340 en sangre de cordón, se recogieron datos somatométricos de los niños (al nacimiento, 18 meses y 4 años) y de las madres (preconcepcional y al parto).
Resultados: Se detectaron Hg y Pb (>2 ng/l) en 329 y 14 recién nacidos respectivamente. El IMC materno preconcepcional fue de 23,8 ±4,3 kg/ m2, la ganancia de peso en el embarazo 13,8 ±5,3 kg., el peso al nacer estandarizado para 40 semanas 3.372 ±397,4 gr, el IMC a los 4 años 16,4 ±1,8 kg/ m2 y la suma de los 4 pliegues (subcutáneo, tricipital, abdominal y suprailíaco) 30,1 ±12,4 mm. 28 niños fueron prematuros y 478 a término. Las primigestas constituían el 61%. No hubo diferencia significativa (p>0,05) para los niveles de Hg como variable continua entre los nacidos pretérmino (13,48 ng/l) y los a término (13,98 ng/l) ni para los de Pb (pretérmino 1,13 ng/l y a término 1,0 ng/l). Tampoco se halló asociación entre dichos niveles en sangre de cordón y el peso al nacer estandarizado para 40 semanas, el IMC, la talla ni con la suma de los 4 pliegues a los 4 años.
Conclusiones: No se encontraron diferencias significativas entre la exposición prenatal al Hg ni al Pb y la somatometría del niño, ni con la prematuridad. Es necesario investigar otros posibles efectos derivados de la exposición a metales pesados durante en la etapa prenatal.
P1/D2-3-011 AUMENTO DEL ÍNDICE DE MASA CORPORAL PREVIO AL INICIO DEL BOTÓN MAMARIO EN PACIENTES ADOPTADAS CON DIAGNÓSTICO DE PUBERTAD PRECOZ /ADELANTADA. F. Carvallo Aita, P. Casano Sancho, L. Suárez, L. Ibañez Toda.
Servicio de Endocrinología Pediátrica. Hospital Sant Joan de Déu, Barcelona.
Introducción: En un estudio realizado recientemente en España se ha constatado que las niñas adoptadas tienen un mayor riesgo de presentar pubertad precoz que las niñas autóctonas. Aunque se desconoce la causa desencadenante se sugiere que aparte de los factores genéticos y psicosociales, la rápida ganancia de peso y cambios nutricionales podrían alterar la maduración en estas niñas.
Objetivo: Valorar la evolución del peso, talla e índice de masa corporal (IMC) en un grupo de niñas adoptadas que desarrollan pubertad precoz o adelantada. Sujetos y Métodos: Estudio retrospectivo en el que se valora a 18 niñas con adopción internacional y pubertad precoz/adelantada, seguidas desde su llegada a nuestro país por la Unidad de Patología Importada del Hospital Sant Joan de Déu. Se estudia su peso, talla, IMC y DE para su edad y sexo, desde su llegada anualmente.
Resultados: La edad de adopción fue a los 4,9 ±2,4 años. El 55% de las pacientes provenían de Asia. La media de peso a la llegada fue de DE -0,99 ± 0,85 y talla DE -0,78 ±1,2. Se registra la aparición del S2 a los 7,3 años (rango 5,2-8,9). La tendencia en el IMC: a su llegada presentaban IMC DE χ= -0,7 ± 0,86, al año χ=-0,12 ± 0,75, a los 2 años =+0,08 ± 0,66 y en la aparición del S2 χ=+0,14 ± 0,59. Se observó una media de aumento en IMC-DE desde su llegada hasta la aparición de S2 de χ=+0,84 ± 0,85. En el 33% de pacientes el IMC en el momento del S2 superaba + 0.5 DE a la media poblacional. El 72% recibió tratamiento con agonistas de GnRH. Seis casos han finalizado pubertad en 35±5,0 meses con menarquia a los 10,4 ± 1,05 años.
Conclusión: 1. Las niñas adoptadas llegan con peso/talla e IMC por debajo de la media poblacional. 2. Existe una tendencia al aumento de peso e IMC en los primeros años desde su adopción 3. Previo al desarrollo del botón mamario aumentan su IMC en cerca de + 1 DE. 4. Es importante llevar un control del crecimiento de estas pacientes para evitar una ganancia de peso rápida y excesiva.
P1/d2-3-012 DISPLASIA INMUNO-ÓSEA DE SCHIMKE Y DÉFICIT DE HORMONA DE CRECIMIENTO. J.M. Donate Legaz(1), JD.González Rodríguez (1), E. Guillén Navarro (2), AP. Nso Roca (1), P. Cortés Mora (1),
(1) Hospital General Universitario Santa Lucía, Cartagena, (2) Unidad Genética Médica. Hospital Universitario Virgen Arrixaca, Murcia.
Introducción: La displasia inmuno-ósea de Schimke (SIOD) es una rara displasia espondiloepifisaria caracterizada por talla baja, nefropatía progresiva, linfopenia, manchas hiperpigmentadas y ateromatosis precoz. Tiene herencia autosómica recesiva describiéndose mutaciones en el gen SMARCAL1.
Caso clínico: Preescolar de 3 años remitido por talla baja con antecedente de consanguinidad, prematuridad con peso y talla pequeños para edad gestacional e hipoglucemia asintomática en periodo neonatal. Exploración física: Peso: 9.9 kg (-2.74 DE) Talla: 78 cm (-5.76 DE) IMC: 15 (-0.8 DE) PC: 46 (-3.34 DE) Aumento de la distancia intermamilar y escroto en chal. Pruebas complementarias: Analítica general normal salvo IGF-1: 17 ng/ml y IGF-BP3 1705 ng/ml. Test de clonidina con pico de GH de 2.7 ng/ml y megatest con insulina, TRH y leuprorelina normal salvo pico de GH de 2.3 ng/ml. RMN: Silla turca parcialmente vacía con neurohipófisis eutópica y craneoestenosis posterior. Evolución: Se inició tratamiento con GH a 0.035 mg/kg/día con incremento de la velocidad de crecimiento durante el primer año de -5.64 DE a tan solo -1.53 DE a pesar de incremento de dosis. Aparición progresiva de distensión abdominal con hiperlordosis lumbar y manchas hiperpigmentadas. A partir del diagnóstico de riñón en herradura en ecografía abdominal se diagnostica de síndrome nefrótico por glomeruloesclerosis segmentaria y focal que se ha comportado como corticorresistente y sin respuesta a inmunosupresores con insuficiencia renal progresiva en la actualidad en estadio IV. Mapa óseo: Con vértebras ovoideas y platispondilia con hipoplasia de pelvis y cavidad acetabular. Aparición de hipotiroidismo subclínico en tratamiento. Ha presentado anemia progresiva, trombocitosis persistente y linfopenia intermitente con inversión del cociente CD4/CD8. Angio RMN normal. En el estudio genético del gen SMARCAL1 se detectó la mutación c.1920-1921insG en homocigosis.
Discusión: A pesar de la existencia de pruebas fehacientes que orienten al diagnóstico de déficit de GH la presencia de talla baja grave con antecedente de pequeño para edad gestacional y la presencia de estigmas en la exploración o en las pruebas complementarias nos debería hacer considerar la posibilidad diagnóstica de las displasias óseas.
P1/d2-3-013 RESPUESTA A HORMONA DE CRECIMIENTO EN PACIENTES CON DÉFICIT DE GH Y RELACIÓN CON TALLA FINAL. M.J. Comesías Gonzalez.
Hospital de Sant Joan de Déu, Martorell (Barcelona).
Objetivo: Revisar la respuesta al tratamiento con hormona de crecimiento recombinante (rGH) de todos los niños deficitarios tratados en nuestro centro en los últimos 20 años y que ya han alcanzado talla final.
Material y Métodos: Se revisan diferentes parámetros, investigando su posible correlación con la talla final. Se valora la edad de inicio del tratamiento y de la pubertad, parámetros antropométricos: talla al inicio del tratamiento, velocidad de crecimiento tras 6 y 12 meses de rGH, incremento de talla en pubertad y talla diana. También se analiza la evolución de la masa ósea y el pico máximo de GH en los test de estimulación así como IGF1 pre y a los 6 m del comienzo de rGH. Se estudia si existe correlación significativa entre algunos de dichos parámetros y la talla final alcanzada. Mediante un análisis de regresión lineal múltiple, se desarrolla un modelo para el cálculo de talla final, seleccionando las variables que se relacionan significativamente con ésta, siguiendo el método stepwise.
Resultados: La muestra comprende un total de 16 niños y 14 niñas. En la tabla se ilustran las medias y DS de los parámetros descriptivos de dicha muestra, que han podido correlacionarse con talla final:
El pico máximo de GH tras estimulación y los niveles pre y post de IGF1 no se correlacionan significativamente con la respuesta al tratamiento.
En el análisis de regresión múltiple, la combinación de variables que se correlaciona mejor con talla final es la talla al inicio de pubertad, las SDS de talla a los 0 y 6 meses de tratamiento, la velocidad de crecimiento a los 6 meses y las SDS de talla al inicio de la pubertad.
Conclusiones: El grado de incremento de talla tras 6 meses de tratamiento con rGH parece ser un buen parámetro precoz de respuesta al tratamiento. La talla al inicio de pubertad tiene también gran interés pronóstico.
P1/d2-3-014 DISCONDROSTEOSIS DE LÉRI-WEILL EN PACIENTE CON DOBLE MUTACIÓN EN HETEROCIGOSIS EN EL GEN SHOX. N. Martín Ruiz (1), A. Cabrejas Lalmolda (1), S. Congost Marín (1), J.I. Labarta Aizpun (1), E. Mayayo Dehesa (1), K. Heath (2).
(1) Hospital Infantil Miguel Servet. Zaragoza, (2) Servicio de Genética Médica Hospital Universitario La Paz. Madrid.
Introducción: La deformidad de Madelung describe una variedad de alteraciones de la muñeca caracterizadas por una fusión prematura de la fisis distal del radio con deformidad consecuente del cúbito distal y muñeca. Se presenta a una paciente con discondrosteosis de Léri-Weill (DLW), cuyo signo guía fue dicha malformación, en la que se ha estudiado el gen SHOX. Se acepta que las mutaciones en heterocigosis en el gen SHOX se asocian con la DLW y las mutaciones en homocigosis o en doble heterocigosis con una displasia de Langer.
Caso: Niña de 9.5 años que presenta deformidad dolorosa de ambas muñecas. Antecedentes familiares: Talla padre: 164,4 cm, talla madre: 157 cm. TH: 154,5 cm. Abuela paterna: 155cm (con segmento inferior corto), abuelo paterno 166 cm. Abuelos maternos de talla normal. Antecedentes personales: Diagnóstico durante la gestación de longitud corta de huesos largos, amniocentesis normal. Parto y periodo neonatal sin incidencias. Peso al nacimiento: 3150 g. (+0,13 SDS). Longitud: 51cm (+0,7 SDS). Desarrollo psicomotor normal.
Exploración física: Talla: 121,9 cm (- 4,94 SDS), segmento inferior 58,5 cm, talla sentada: 71,2cm, envergadura: 110,5cm. Cúbito valgo. No hábito Turner. Extremidades superiores mesomélicas. Peso: 28,6 (-0,78 SDS). Estadío puberal: Tanner I. Pruebas complementarias: Bioquímica y función tiroidea normales. IGF-1: 30 ng/ml. Resto de estudio hormonal normal. Edad ósea correspondiente a su edad. Cariotipo: 46XX. Estudio del gen SHOX y la región PAR1 (pseudoautosomal region1) mediante MPLA (multiplex ligation-dependent probe amplification): Deleción en heterocigosis que cubre la totalidad del gen SHOX desde la sonda L10292 a la sonda L05105 de PAR1 en el flanco 3’ distal de SHOX y deleción de las sondas L05099 a L05101 en el otro alelo.
Comentario: Se presenta un caso de discondrosteosis de Léri-Weill con deleción en doble heterocigosis del gen SHOX en el que clínicamente se ha descartado una displasia de Langer.
P1/d2-3-015 ANÁLISIS COMPARATIVO DE COSTES ENTRE LAS DISTINTAS PRESENTACIONES DE HORMONA DE CRECIMIENTO: JERINGAS MONODOSIS VS VIALES MULTIDOSIS. A. Mas Comas(1), M.G. López Ramos (1), M. Villaronga Flaqué (1), A. Murciano Cabeza (1), S. Díaz Cerezo (2),
(1) Servicio de Farmacia. Hospital Sant Joan de Déu. Barcelona; (2) Departamento de Investigación de Resultados en Salud, Pfizer, Esplugues de Llobregat (Barcelona). Objetivo: Realizar un análisis de costes comparativo entre las distintas presentaciones de hormonas de crecimiento (GH).
Métodos: Estudio observacional retrospectivo a partir del registro de GH del Servicio de Farmacia de un hospital universitario materno-infantil. Se diseñó un análisis de costes comparativo entre las distintas presentaciones de GH (jeringas monodosis vs. viales multidosis). Se incluyeron pacientes que iniciaron el tratamiento con GH entre Enero 2006 y Diciembre 2009 y que hubiesen retirado medicación durante mínimo 6 meses. Se calcularon los mg perdidos de medicación (=mg dispensados-mg teóricos según posología) anuales y su coste medio anual/ paciente (según PVL). Se calculó también el porcentaje de incumplidores (recogían mínimo un 8% menos de la dosis prescrita) y el porcentaje de pacientes con ajuste de dosis perfecto (±1% pérdidas)
Resultados: 306 pacientes cumplieron los criterios de inclusión; edad inicio tratamiento 8,2 (3,6); 51,0% hombres. 257 (84%) fueron válidos para el análisis de pérdidas. Los restantes 49 (16%) recogieron menos medicación de la prescrita y de éstos, 19 (6,3%) fueron incumplidores. Los pacientes incluidos en el análisis de pérdidas fueron homogéneos en las variables sexo, edad de inicio tratamiento, duración del tratamiento e indicación en ambos grupos; p>0.05. La siguiente tabla resume los resultados:
Conclusión: Las pérdidas de GH de las distintas presentaciones no superan en ningún caso el 10% de la dosis dispensada. El uso de jeringas monodosis supuso un menor porcentaje de perdidas anuales y un mayor número de pacientes recibió la dosis justa según lo prescrito.
P1/d2-3-016 HIPERCRECIMIENTO EN GEMELAS. A. De La Puente Arévalo(1), E. Bermúdez de Castro López (1), AC. Barreda Bonis (1), L. Salamanca Fresno (1), P. Lapunzina (2), R. Gracia Bouthelier (1).
(1) Servicio de Endocrinología Infantil, Hospital Universitario La Paz, Madrid, (2) Instituto de Genética Médica y Molecular (INGEMM), Hospital Universitario La Paz, Madrid.
Dentro de los síndromes de hipercrecimiento el síndrome de Bannayan-Riley-Ruvalcaba (SBRR) es una rara entidad genética autosómica dominante ocasionada en un 60% de los casos por mutaciones en el gen homólogo de fosfatasa y tensina delecionado del cromosoma 10 (gen PTEN) implicado en la supresión tumoral. Caracterizado por macrocefalia, retraso psicomotor, hamartomas intestinales, lipomatosis, hemangiomatosis y máculas pigmentadas en el pene. Presentamos a dos pacientes mujeres de 9 meses de edad, producto de una gestación gemelar espontánea con parto a las 35 semanas. Respecto a la antropometría al nacimiento la primera gemela presentó un peso en + 0.78 DE, una longitud en De los antecedentes familiares destacar que ambos padres son sanos, no consanguíneos, con estatura normal. Tienen dos hermanos mayores altos. En la exploración física al nacimiento destaca en ambas a nivel craneofacial: macrocefalia, frente alta, hipotelorismo, epicantus, estrabismo divergente, raíz nasal deprimida, cuello corto y paladar ojival. Neurológicamente reseñar hipotonía axial. Ambas son valoradas por neurocirugía: la primera gemela presenta una escafocefalia y precisa corrección quirúrgica, la segunda únicamente precisa medidas posturales. Ambas presentan un retraso en el desarrollo psicomotor. Sospechando un cuadro sindrómico se completa el estudio con examen cardiológico detectándose CIA en la primera gemela siendo el estudio normal en la segunda, ecografía abdominal y cerebral normales. En el estudio genético ambas son portadoras en heterocigosidad de la deleción c.210-7 del CTTTT en el gen PTEN. Se diagnostican de SBRR. En el SBRR se describe una longitud aumentada al nacimiento con talla adulta normal y perímetro cefálico aumentado toda la vida. Presentan pólipos intestinales. Neurológicamente destaca hipotonía, convulsiones y retraso psicomotor. Existe mayor riesgo de enfermedades autoinmunes. Los pacientes con síndromes de tumores hamartomatosos por mutación del PTEN tienen riesgo aumentado de cáncer. El diagnóstico genético ante la sospecha de estos síndromes es fundamental a la hora de decidir realizar un screening de tumores malignos y estudio genético familiar.
P1/d2-3-017 ESTUDIO SOBRE LA EVOLUCIÓN DE IGF1, IGF-BP3 Y EL COCIENTE ENTRE AMBOS EN NIÑOS TRATADOS CON HORMONA DE CRECIMIENTO. E. Bermúdez de Castro López, A. De La Puente Arévalo, J. Guerrero-Fdez, L. Salamanca Fresno, I. González Casado, R. Gracia Bouthelier.
Servicio de Endocrinología Infantil, Hospital Universitario La Paz, Madrid.
Introducción: Para monitorizar el tratamiento con Hormona de Crecimiento (GH) y evitar posibles efectos secundarios, se recomienda la medición regular de IGF1 e IGFBP3. Los niveles de IGF1 en el límite superior y de IGFBP3 en el inferior se relacionan con mayor riesgo de desarrollo de neoplasias.
Objetivos: Estudio de la evolución a largo plazo de los niveles de IGF1, IGFBP3 y el cociente entre ambos en niños tratados con GH.
Material y Métodos: 82 pacientes (20 con síndrome de Turner, 21 con déficit idiopático de GH, 20 con déficit orgánico y 21 con retraso de crecimiento intrauterino (RCIU)), que fueron tratados con GH entre tres y cinco años.
Resultados: Se observa un aumento significativo en todos los grupos de las cifras de IGF1 e IGFBP3 a lo largo del tratamiento, siendo este aumento mayor en los pacientes con Turner y menor en el déficit orgánico. Las medias de IGF1 e IGFBP3 al inicio y a los 5 años de seguimiento respectivamente en cada grupo son las siguientes:
- Turner. IGF1: 169,76 y 573,2 ng/ml; IGFBP3: 3,38 y 6,5 mcg/dl - Déficit idiopático. IGF1: 93,67 y 398,87 ng/ml; IGFBP3: 2,75 y 5,93 mcg/dl - Déficit orgánico. IGF1: 41,75 y 323,7 ng/ml; IGFBP3: 1,5 y 4,8 mcg/dl - RCIU. IGF1: 107,81 y 556 ng/ml; IGFBP3: 2,44 y 6,7 mcg/dl. Además, se produce una elevación inicial del cociente IGF1/IGFBP3 con una estabilización al final del seguimiento, manteniéndose en todos los grupos entre 50 y 100.
Conclusiones: Hasta ahora no se ha demostrado una relación clara entre el tratamiento con GH y el desarrollo de neoplasias en ausencia de factores de riesgo. Sin embargo, la Growth Hormone Research Society recomienda la monitorización de IGF1 e IGFBP3 en estos pacientes. No se ha determinado una cifra concreta de IGF1/IGFBP3. En vista de los resultados de este estudio, recomendamos mantener este cociente entre 50 y 100, ya que es la cifra en la que se mantienen no solo los pacientes con déficit orgánico de GH (en los que este déficit es más evidente), sino también en los otros tres grupos estudiados.
P1/d2-3-018 COEXISTENCIA DE 2 TRASTORNOS RECESIVOS EN UNA MISMA PACIENTE: SINDROME 3-M Y DÉFICIT DE METILACETOACETATO HIDROLASA. A. González Jimeno(1), A. Carcavilla Urqui (1) , F. López Grondona (1), O. Garcia Campos (1), D. Hanson (2),
(1) Hospital Virgen de la Salud. Toledo; (2) Manchester Academic Health Sciences Centre, Manchester.
Introducción: El síndrome 3-M (OMIM #273750) es un trastorno autosómico recesivo caracerizado por retraso del crecimiento intrauterino y postnatal, rasgos dismórficos, macrocefalia y alteraciones óseas. Se ha descrito asociado a mutaciones en los genes CUL7 y OBSL1. El déficit de metilacetoacetato tiolasa (MAT; OMIM 203750) es un defecto congénito del metabolismo de transmisión autosómica recesiva, que afecta el catabolismo de los cuerpos cetónicos. Clínicamente, se caracteriza por episodios de vómitos, deshidratación, cetoacidosis y depresión sensorial que pueden evolucionar a coma. Se debe a mutaciones en el gen ACAT1. Presentamos un caso de coexistencia de las 2 enfermedades en una familia marroquí consanguínea.
Caso clínico: Niña nacida a término ingresa al nacimiento por CIR severo y rasgos dismórficos. A la exploración destacan: Peso 1.740 g (-2,66 DS), longitud: 39,5 cm (-4,38 DS) y perímetro cefálico (PC): 34 cm (0,29 DS). Macrocefalia relativa de configuración dolicocéfala, labios carnosos, tórax corto, abdomen prominente, hiperlaxitud de miembros y talones prominentes. Padres consanguíneos (primos hermanos) sanos. Hermano, seguido en nuestra consulta por retraso del crecimiento de origen prenatal y rasgos dismórficos similares, con talla en -3,97 DE a los 2 años 5 meses de edad. A los 6 meses de vida la paciente ingresa por sospecha de sepsis acompañada de acidosis metabólica grave. Ante la sospecha de metabolopatía se inicia estudio bioquímico con el resultado de aumento de metabolitos de cetosis. El estudio enzimático en fibroblastos determina una actividad de Acetoacetil-coA Tiolasa deficiente, lo que indica un defecto de la cetolisis a nivel de la actividad de
Métodos: Secuenciación del gen CUL7 en la paciente y hermano, y del gen ACAT1 en la paciente.
Resultados: El hermano y el caso índice son homocigotos para una mutación conocida en CUL7: c.4451_4452delTG p.V1484GfsX68. Pendiente del estudio genético de MAT de la paciente.
Conclusiones: Se trata, hasta donde sabemos, del primer caso descrito hasta la fecha de coexistencia de estos 2 trastornos heredados de manera recesiva, y uno de los pocos casos descritos de síndrome 3M en nuestro medio. El pronóstico a largo plazo estará marcado por la metabolopatía y sus descompensaciones en el futuro.
P1/d2-3-019 SÍNDROME DE LA TRÍADA HIPOTALÁMICA. DESCRIPCIÓN DE UNA SERIE DE CASOS. M.J. Olmos Jiménez, M. Royo Gómez, B. Huidobro Fernández, M. A. Gil-Ruiz Gil Esparza, L. González Vives, A. Rodríguez Sánchez.
Hospital General Universitario Gregorio Marañón, Madrid.
Introducción: El síndrome de tríada hipotalámica (hipoplasia o agenesia de adenohipófisis, ausencia de tallo hipofisario y neurohipófisis ectópica/ausente) es un hallazgo en la RMN que aparece con frecuencia en casos de hipopituitarismo múltiple, aunque también lo encontramos en pacientes con déficit aislado de GH. Éstos últimos pueden desarrollar diferentes déficit hormonales a lo largo de los años. Con el objetivo de conocer la frecuencia, edad y modo de aparición de los déficit hormonales asociados al síndrome de la tríada hemos realizado un estudio descriptivo en los pacientes con diagnóstico de déficit de GH confirmado.
Sujetos y Métodos: Estudio descriptivo retrospectivo en pacientes menores de 16 años diagnosticados de tríada hipotalámica por RMN cerebral. Variables epidemiológicas recogidas: edad, sexo, antecedentes personales y familiares, clínica, datos auxológicos, alteraciones hormonales, tratamiento y evolución mediante revisión de historias clínicas.
Resultados: Muestra de 20 pacientes (N=20) con tríada hipotalámica en RMN La edad media al diagnóstico fue de 3.02 [1.58-5.71] años. El 60% fueron varones. El 70% consultaron por talla baja, con talla y peso al diagnóstico -2.85 [-3.75-(-2.46)] y -1.69 [1.99- El 35% presentó déficit de ACTH, el 55% déficit de TSH y el 100% déficit de GH. Todos recibieron tratamiento hormonal sustitutivo: Hidrocortisona 13-20 mg/m2 oral, L-Tiroxina 0,7-7,6 mcg/kg/día oral, GH 0.027-0.04 mg/kg/día s.c. La velocidad de crecimiento aumentó de -1.8 [-2,64-(-1,0)] a +3.68 [+2,04-(+6,24)] DS al año de tratamiento con GH.
Conclusiones: La realización de RMN cerebral es fundamental ante el hallazgo de un déficit de GH. En pacientes con síndrome de tríada hipotalámica debe realizarse seguimiento a largo plazo de la función hipotálamo-hipofisaria, ya que pueden asociar otros déficit hormonales durante su evolución. En nuestra serie el déficit más frecuente fue el de GH seguido por los de TSH y ACTH.
P1/d2-3-020 REVISIÓN DE LAS ALTERACIONES DEL GEN SHOX EN EL SÍNDROME DE LÉRI-WEILL Y TALLA BAJA IDIOPÁTICA EN MALLORCA. M. Soriano Arola(1), M. Caimari Jaume (1), D. de Sotto Esteban (1), J. Rosell Andreo (2), D. Heine Suñer (2).
(1) Servicio de Pediatría Hospital Son Espases. Palma de Mallorca; (2) Servicio de Genética. Hospital Universitario Son Espases. Palma de Mallorca.
Introducción: Estos últimos años se han implicado las alteraciones del gen SHOX (short stature homeobox) localizado en las regiones pseudoautosómicas de ambos cromosomas sexuales con diversos fenotipos de talla baja que van desde la talla baja idiopática (TBI) al síndrome de Léri-Weill (SLW), que asocia talla baja con deformidad de Madelung.
Objetivos: Presentamos una revisión de los casos de alteraciones del gen SHOX detectadas en familias con talla baja de Mallorca, remitida de diversos hospitales (Hospital Son Dureta, Son Espases y Hospital Son Llàtzer), en los últimos 10 años (2000-2010). Nuestro objetivo es averiguar la relación entre las alteraciones encontradas de dicho gen y el fenotipo que presentan los pacientes.
Resultados: Se han encontrado 17 familias con alteraciones del gen SHOX: 9 delecciones, 4 mutaciones, 3 duplicaciones y 1 delección-duplicación. La mayoría de los pacientes presentaban SLW y/o deformidad de Madelung (64 %), el resto de casos presentaban TBI. En los casos con SLW y/o deformidad de Madelung se encontraron 8 delecciones, 2 mutaciones y 1 delección-duplicación; y entre los casos de TBI 3 duplicaciones, 2 delecciones y 1 mutación. En los pacientes que cumplieron criterios para tratamiento con somatotropina se inició dicho tratamiento presentando una evolución clínica favorable.
Conclusiones: En nuestra revisión hemos observado alteraciones del gen SHOX no sólo en SLW y/o deformidad de Madelung, sino también en pacientes con TBI en una proporción similar a la referida en la bibliografía (2-15 %). Destacar la importancia de realizar estudio genético en pacientes con TBI. El diagnóstico genético junto con la antropometría puede beneficiar al paciente al iniciarse tratamiento con somatotropina. En los casos de SLW, deformidad de Madelung o tallas bajas que asocien algún otro rasgo dismórfico la frecuencia de alteraciones de dicho gen es mayor. Todavía no hemos podido establecer una clara relación entre el tipo de alteración del gen SHOX y el fenotipo presente en los pacientes. |
| References |
 |
 |
 |
 |
 |
 Print
Print View / Download PDF
View / Download PDF  English version
English version Comments
Comments Add Favorites
Add Favorites